10X Visium kidney dataset
0. Environment
library(Giotto)
# STOP!
# ====== 1. set working directory ======
# For Docker, set my_working_dir to /data
my_working_dir = '/data'
# For native install users, set my_working_dir to a directory accessible by user
#my_working_dir = "/home/qzhu/Downloads"
# ====== 2. set giotto python path ======
# For Docker, set python_path to /usr/bin/python3
python_path = "/usr/bin/python3"
# For native install users, python path may be different, and may depend on whether conda or virtual environment. One can check by "reticulate::py_discover_config()" to see which python is picked up automatically. Then set python_path to what is returned by py_discover_config.
# python_path = "/usr/bin/python3"
STOP!
If this is your first time using Giotto after installing Giotto natively, you might want to check you have the environment and pre-requisite packages in python and R installed. Note: this is not relevant to Docker users because it already includes all pre-requisites.
If you are using Giotto in Docker, please see Docker file directories about organization of files in Docker (dataset files, sharing of files between guest and host).
1. Dataset preparation steps
1.1. Dataset explanation
10X genomics recently launched a new platform to obtain spatial expression data using a Visium Spatial Gene Expression slide.
The Visium kidney data to run this tutorial can be found here
Visium technology:
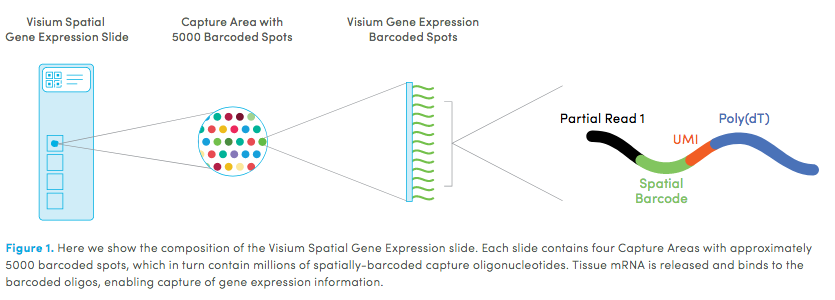
High resolution png from original tissue:
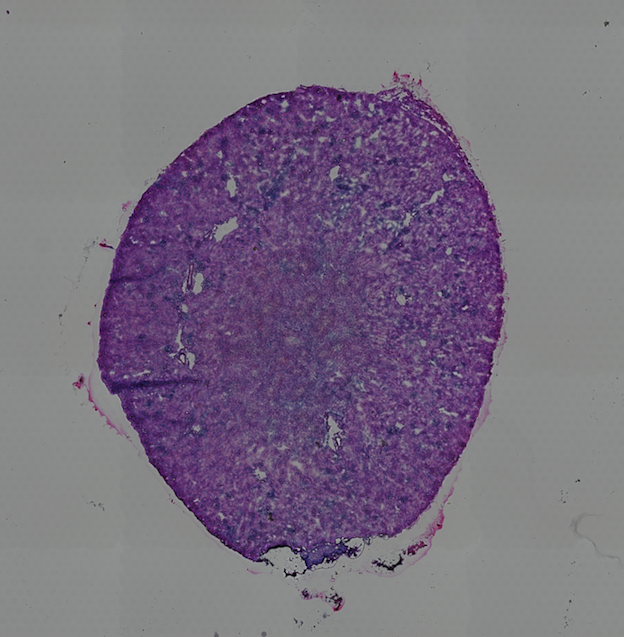
1.2. Dataset download
Due to licensing restrictions, we cannot directly link the dataset here. You need to go to 10X visium dataset page to register an user account. Then click within the dataset, download the link "Feature / cell matrix (raw)" (42.06MB). Then extract the tar.gz file. You will see the directory "raw_feature_bc_matrix" created.
You will also need to download the "Spatial imaging data" (7.62MB). Then extract the tar.gz file. You will see the folder "spatial" created, within which you will find "tissue_positions_list.csv".
1.3. Giotto global instructions and preparations
## create instructions
instrs = createGiottoInstructions(save_dir = my_working_dir,save_plot = TRUE,show_plot = FALSE,python_path = python_path)
## provide path to visium folder
data_path = my_working_dir #containing raw_feature_bc_marix and spatial_folders
2. Create Giotto object & process data
## directly from visium folder
visium_kidney = createGiottoVisiumObject(visium_dir = data_path, expr_data = 'raw',png_name = 'tissue_lowres_image.png',gene_column_index = 2, instructions = instrs)
## update and align background image
# problem: image is not perfectly aligned
spatPlot(gobject = visium_kidney, cell_color = 'in_tissue', show_image = T, point_alpha = 0.7,save_param = list(save_name = '2_a_spatplot_image'))
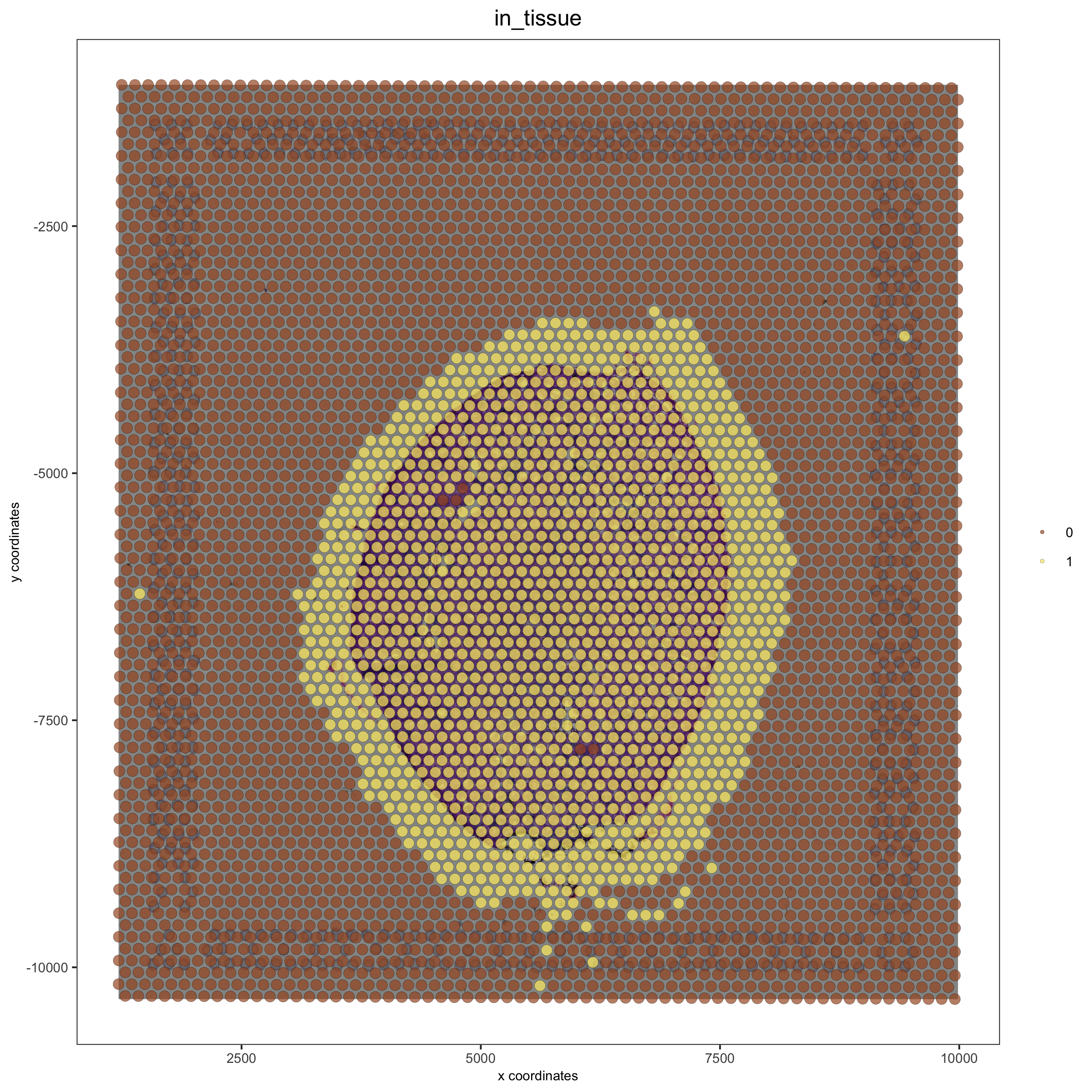
# check name
showGiottoImageNames(visium_kidney) # "image" is the default name
# adjust parameters to align image (iterative approach)
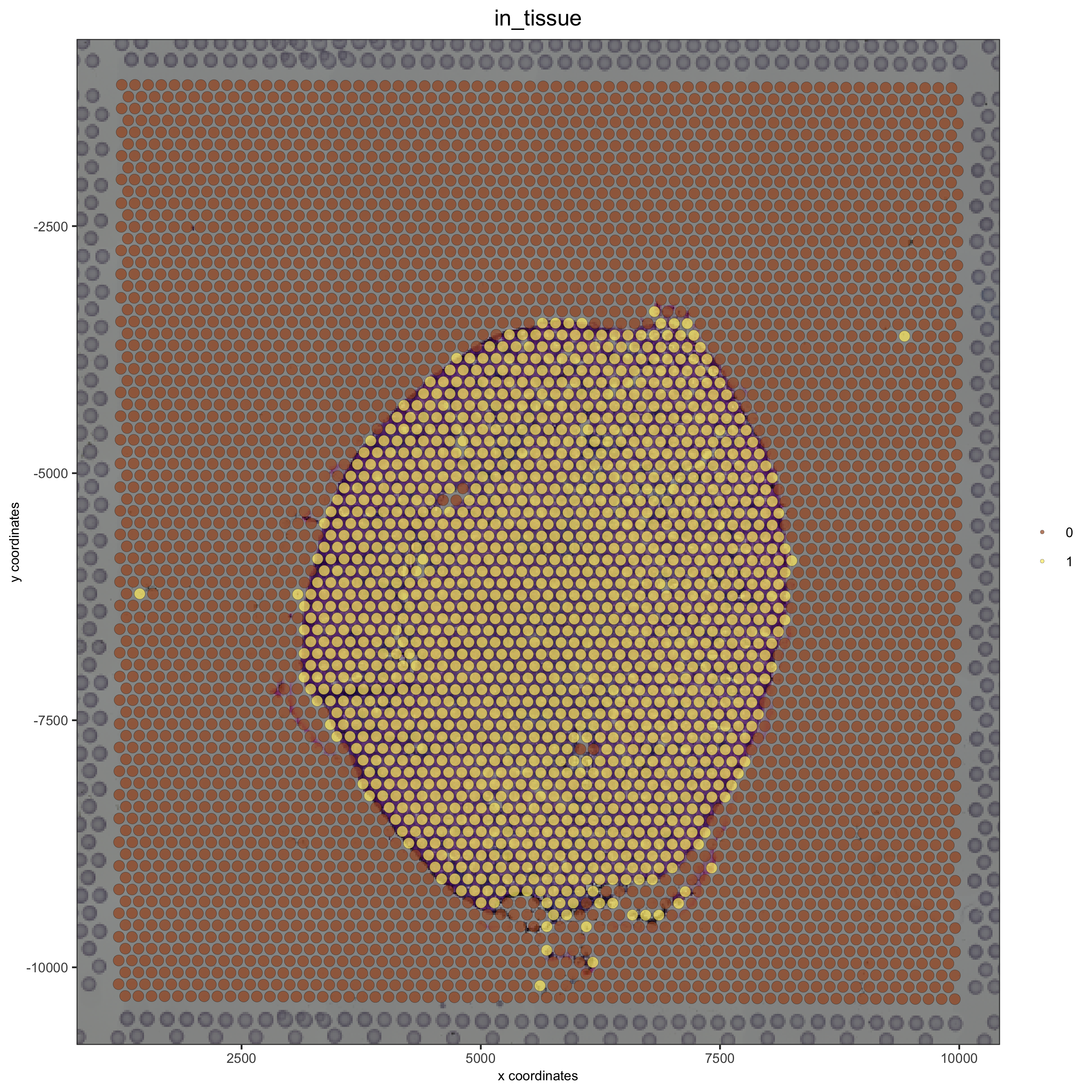
visium_kidney = updateGiottoImage(visium_kidney, image_name = 'image',xmax_adj = 1300, xmin_adj = 1200,ymax_adj = 1100, ymin_adj = 1000)
# now it's aligned
spatPlot(gobject = visium_kidney, cell_color = 'in_tissue', show_image = T, point_alpha = 0.7,save_param = list(save_name = '2_b_spatplot_image_adjusted'))
## check metadata
pDataDT(visium_kidney)
## compare in tissue with provided jpg
spatPlot(gobject = visium_kidney, cell_color = 'in_tissue', point_size = 2,cell_color_code = c('0' = 'lightgrey', '1' = 'blue'),save_param = list(save_name = '2_c_in_tissue'))
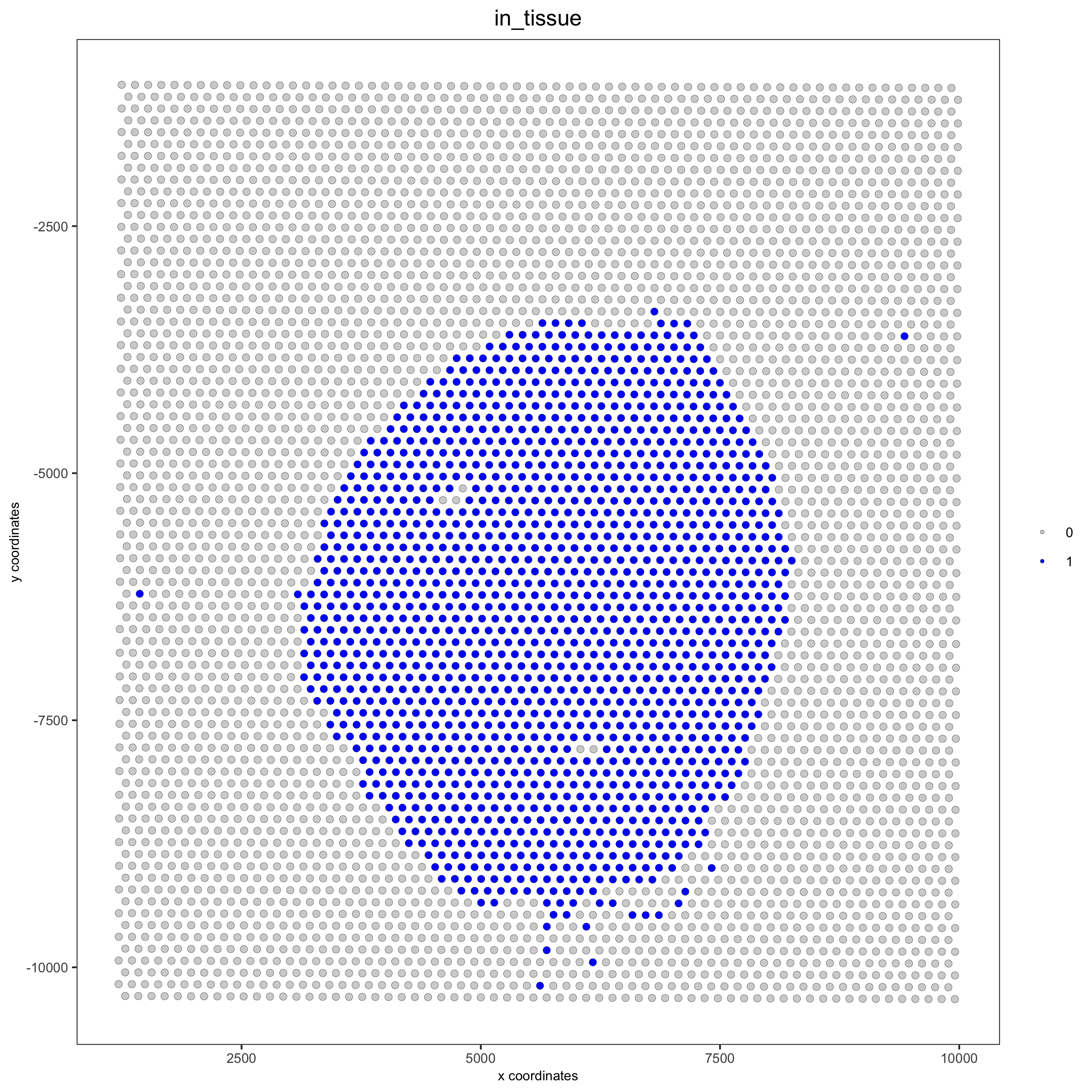
## subset on spots that were covered by tissue
metadata = pDataDT(visium_kidney)
in_tissue_barcodes = metadata[in_tissue == 1]$cell_ID
visium_kidney = subsetGiotto(visium_kidney, cell_ids = in_tissue_barcodes)
## filter
visium_kidney <- filterGiotto(gobject = visium_kidney,expression_threshold = 1,gene_det_in_min_cells = 50,min_det_genes_per_cell = 1000,expression_values = c('raw'),verbose = T)
## normalize
visium_kidney <- normalizeGiotto(gobject = visium_kidney, scalefactor = 6000, verbose = T)
## add gene & cell statistics
visium_kidney <- addStatistics(gobject = visium_kidney)
## visualize
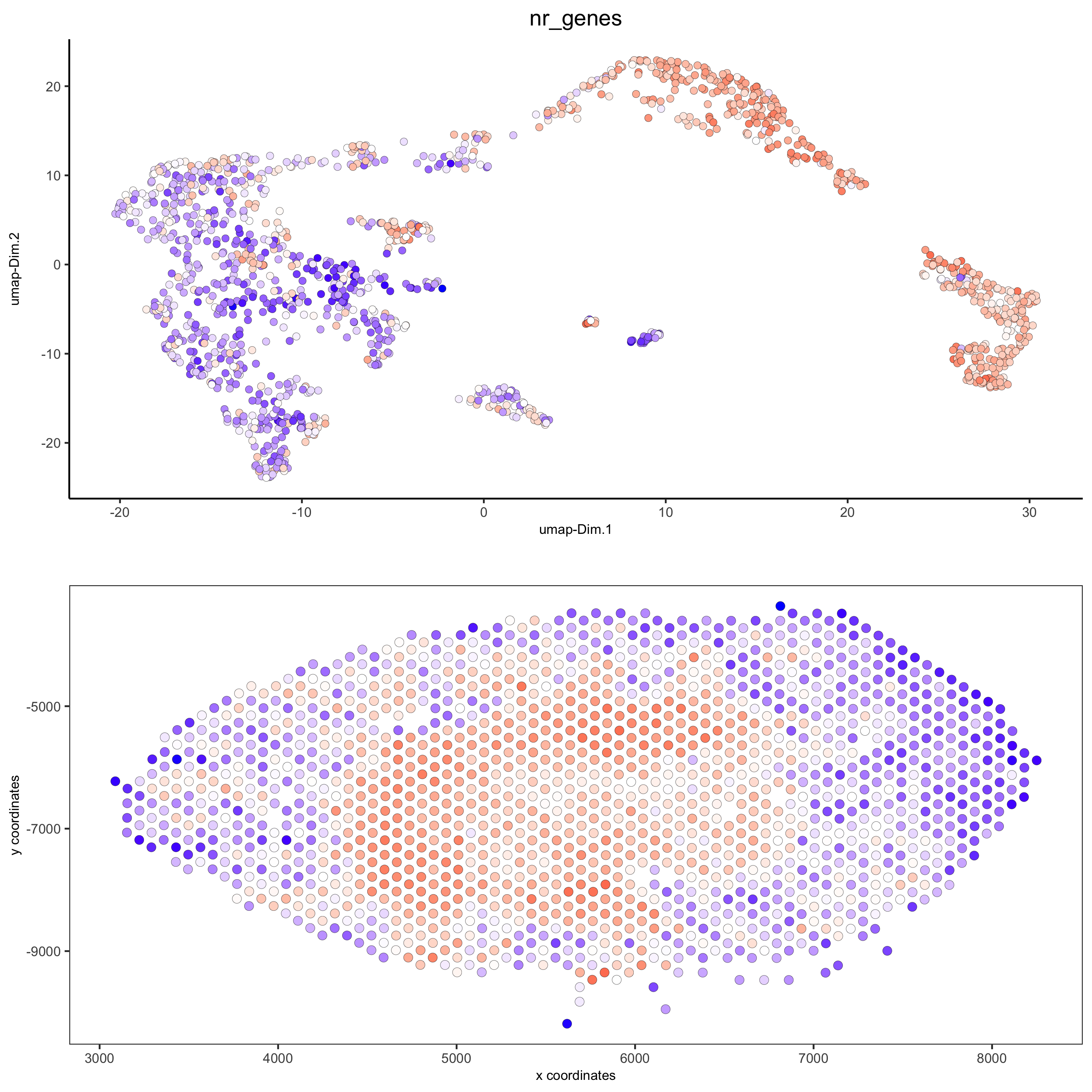
spatPlot2D(gobject = visium_kidney, show_image = T, point_alpha = 0.7,save_param = list(save_name = '2_d_spatial_locations'))

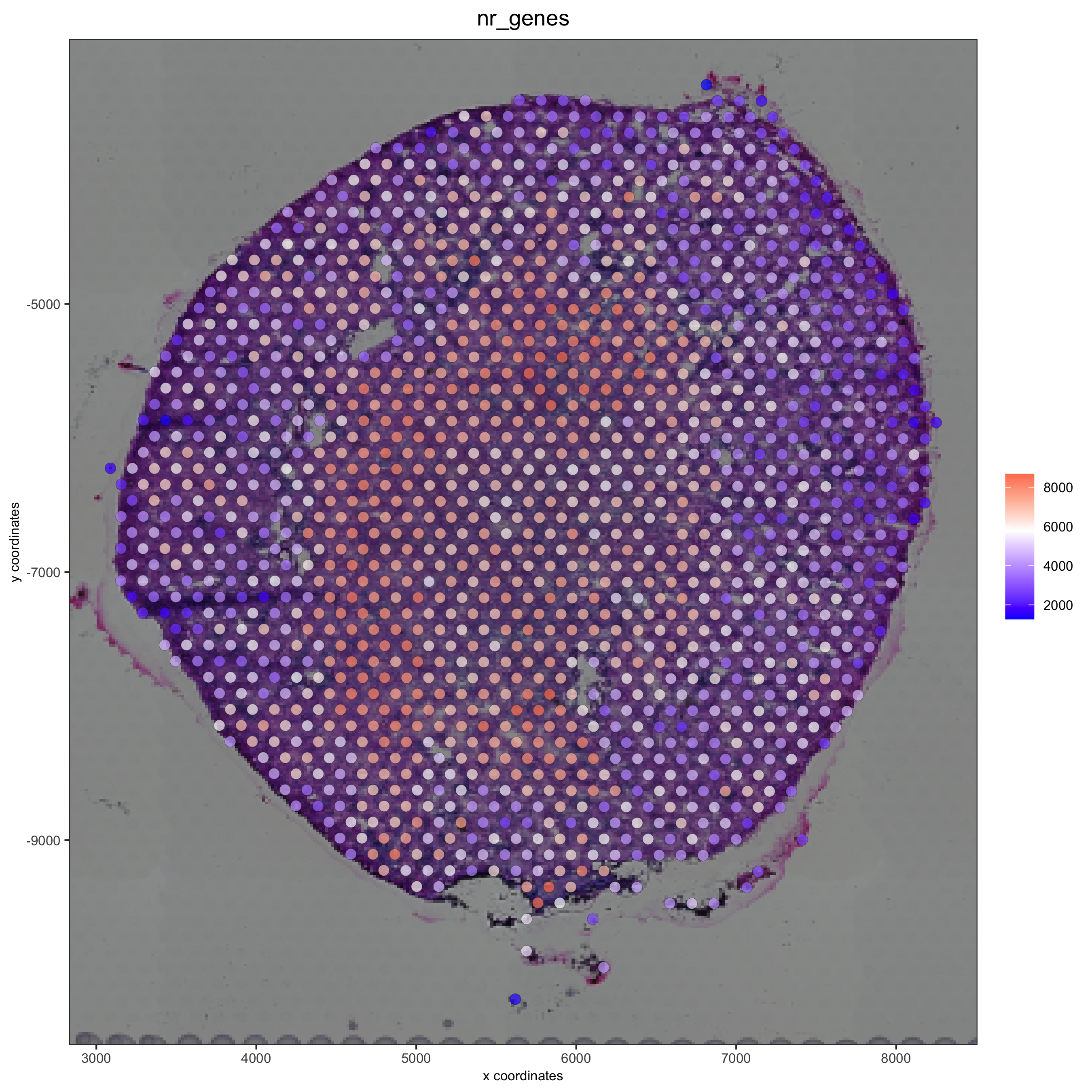
spatPlot2D(gobject = visium_kidney, show_image = T, point_alpha = 0.7,cell_color = 'nr_genes', color_as_factor = F,save_param = list(save_name = '2_e_nr_genes'))
3. Dimension reduction
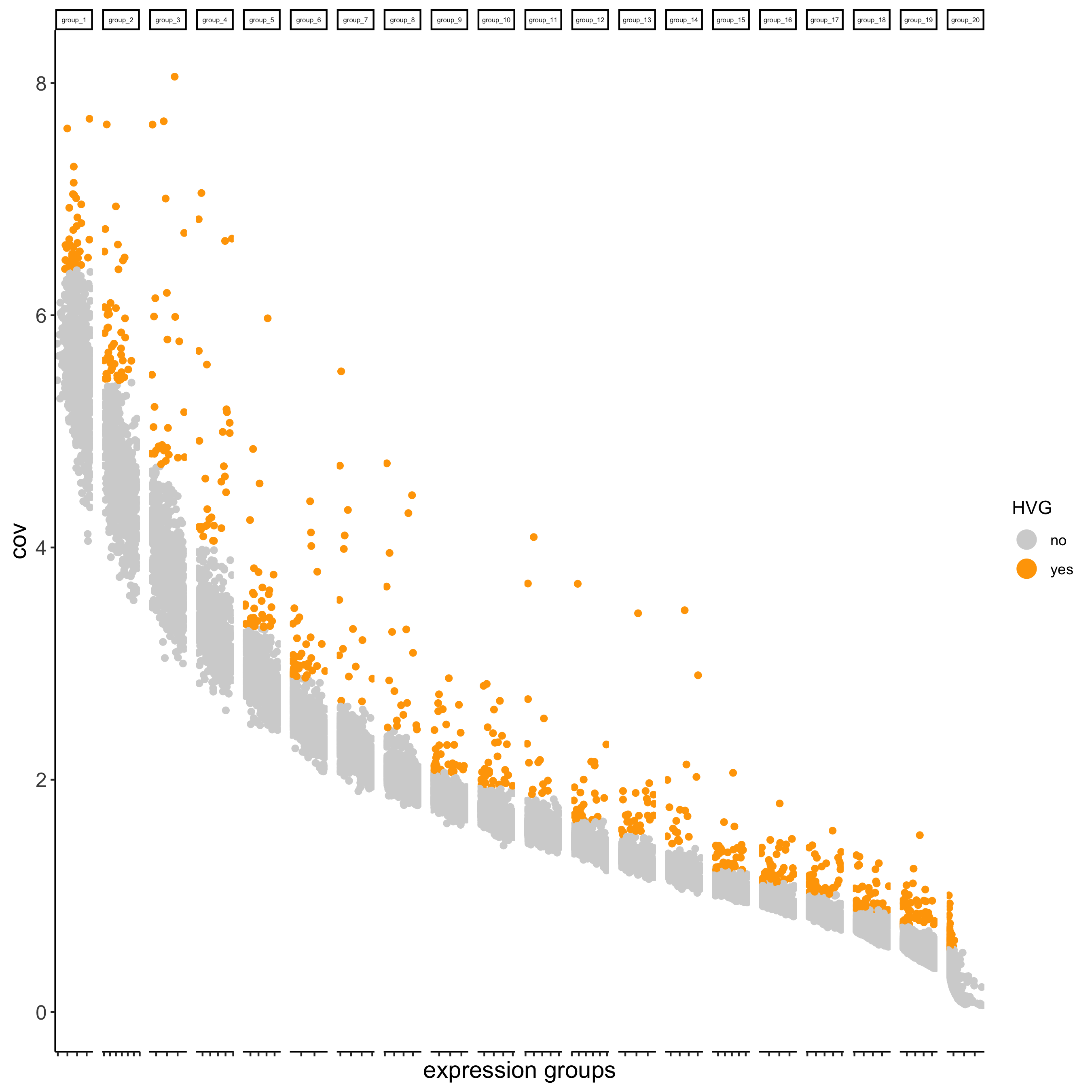
## highly variable genes (HVG)
visium_kidney <- calculateHVG(gobject = visium_kidney,save_param = list(save_name = '3_a_HVGplot'))
## run PCA on expression values (default)
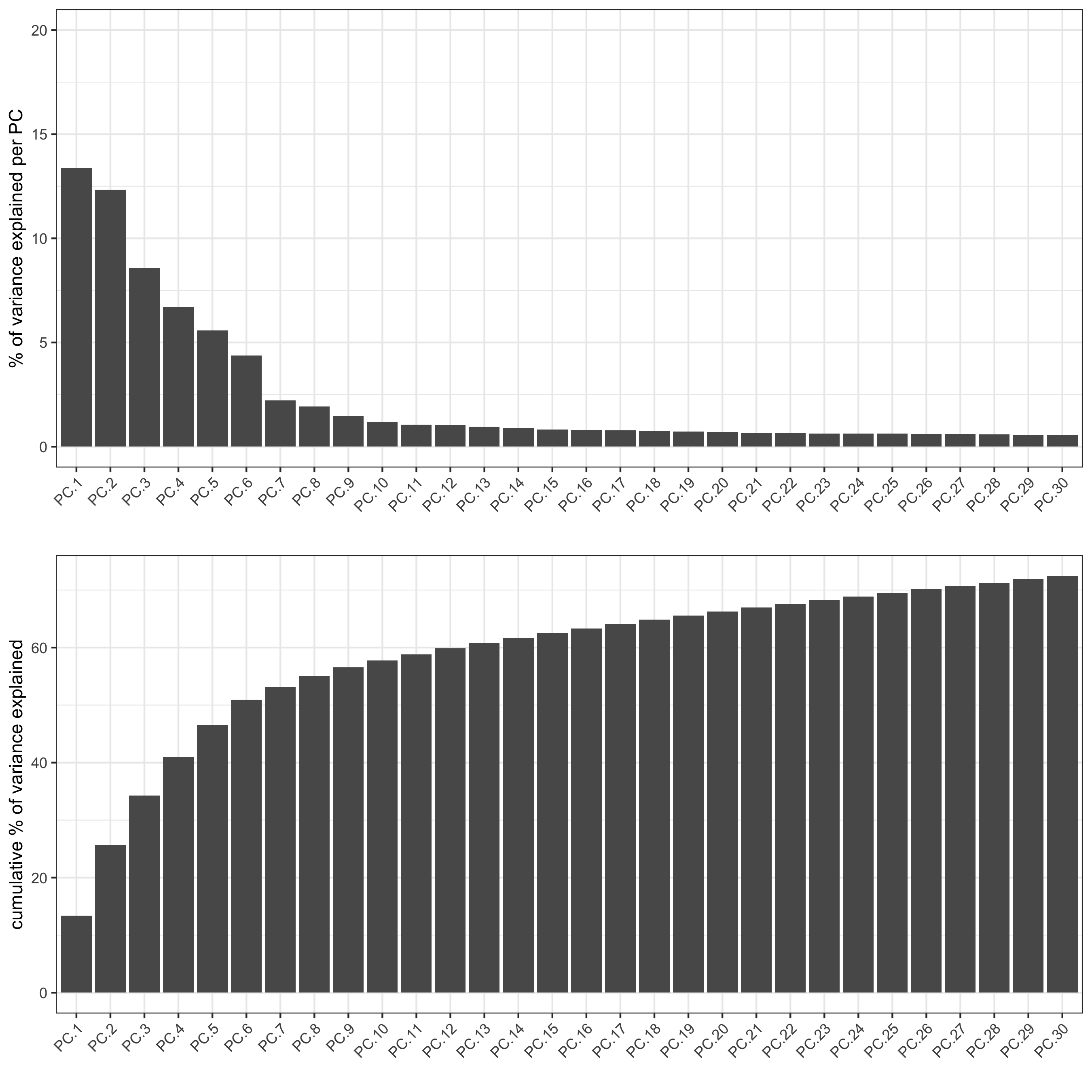
visium_kidney <- runPCA(gobject = visium_kidney, center = TRUE, scale_unit = TRUE)
screePlot(visium_kidney, ncp = 30, save_param = list(save_name = '3_b_screeplot'))
plotPCA(gobject = visium_kidney,save_param = list(save_name = '3_c_PCA_reduction'))
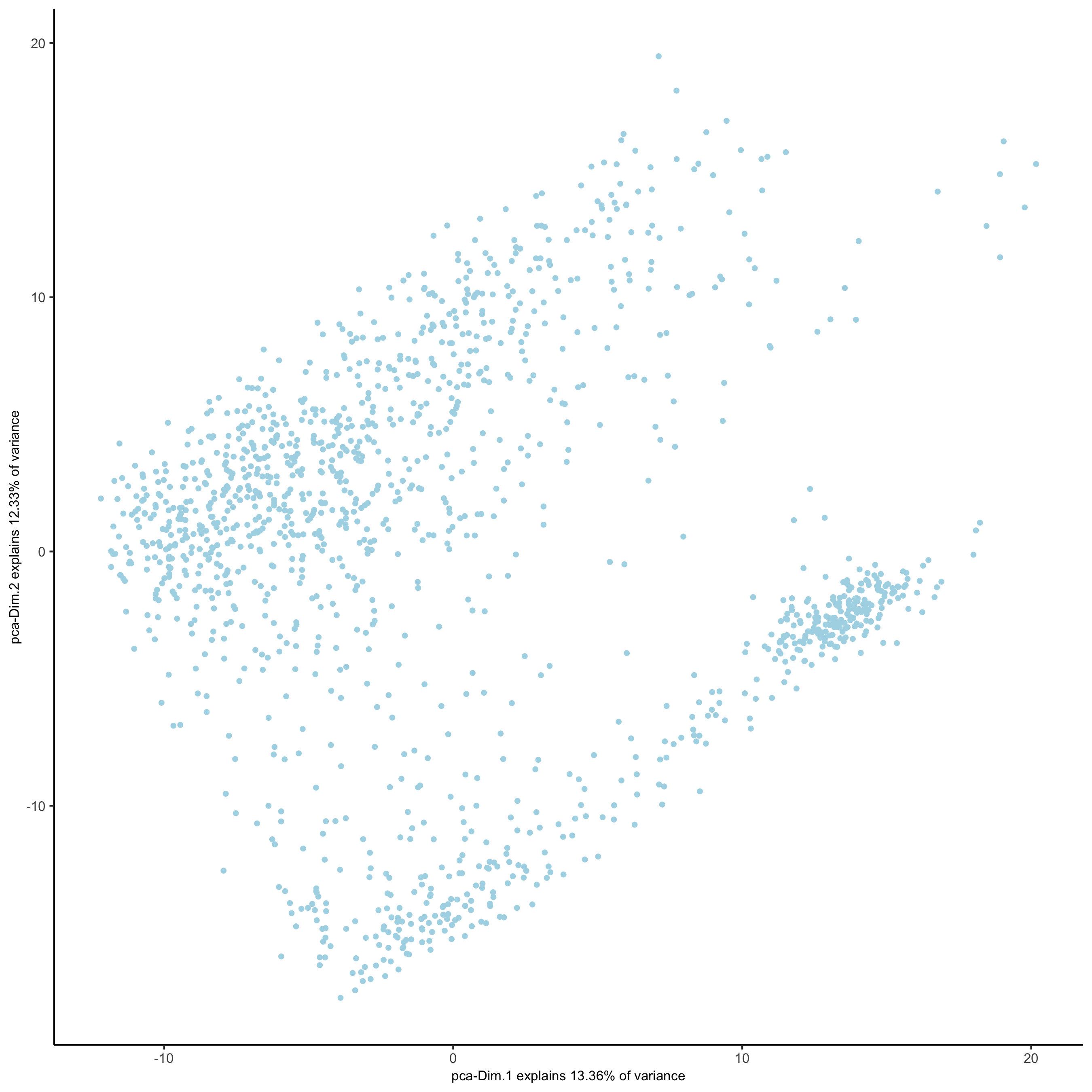
## run UMAP and tSNE on PCA space (default)
visium_kidney <- runUMAP(visium_kidney, dimensions_to_use = 1:10)
plotUMAP(gobject = visium_kidney,save_param = list(save_name = '3_d_UMAP_reduction'))
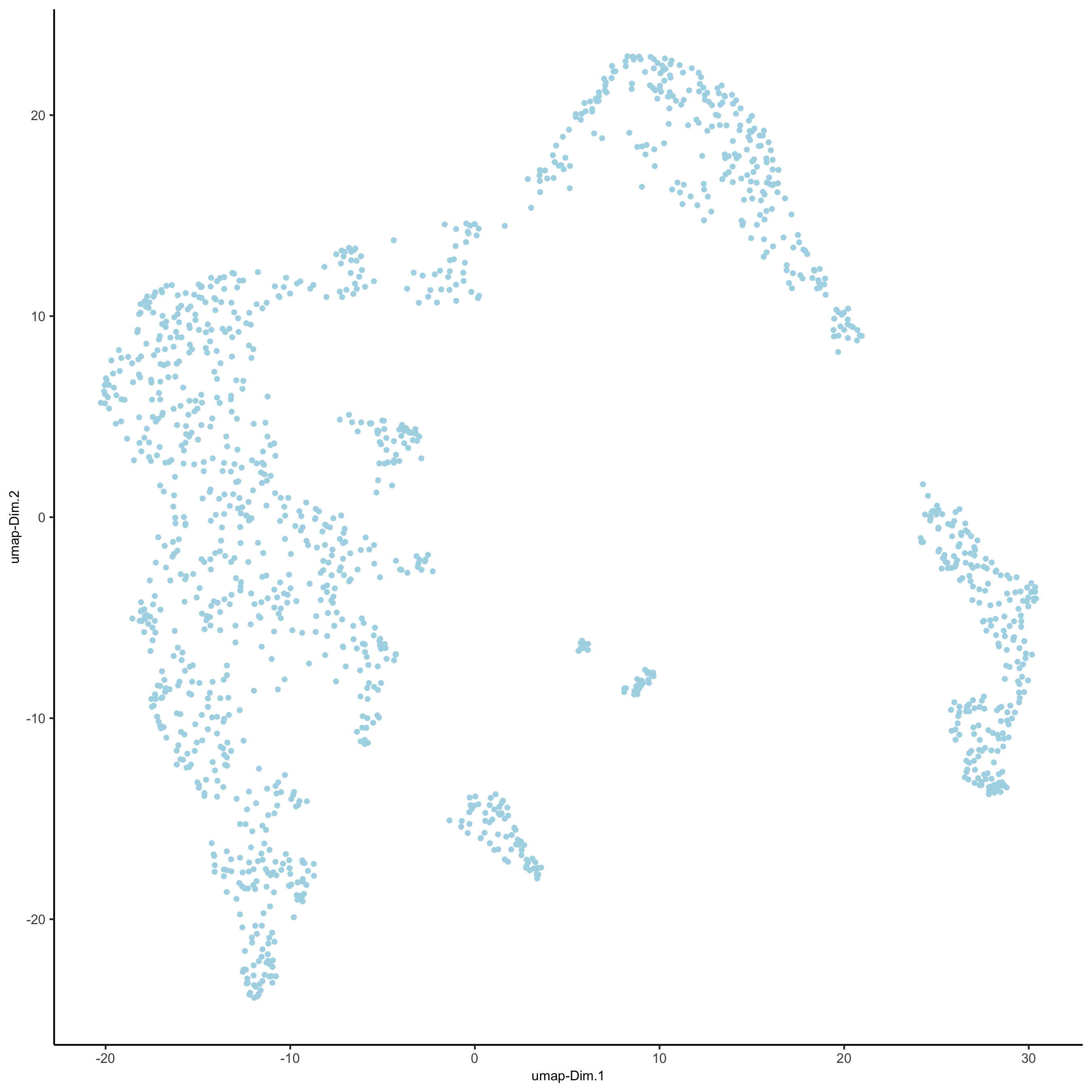
visium_kidney <- runtSNE(visium_kidney, dimensions_to_use = 1:10)
plotTSNE(gobject = visium_kidney,save_param = list(save_name = '3_e_tSNE_reduction'))
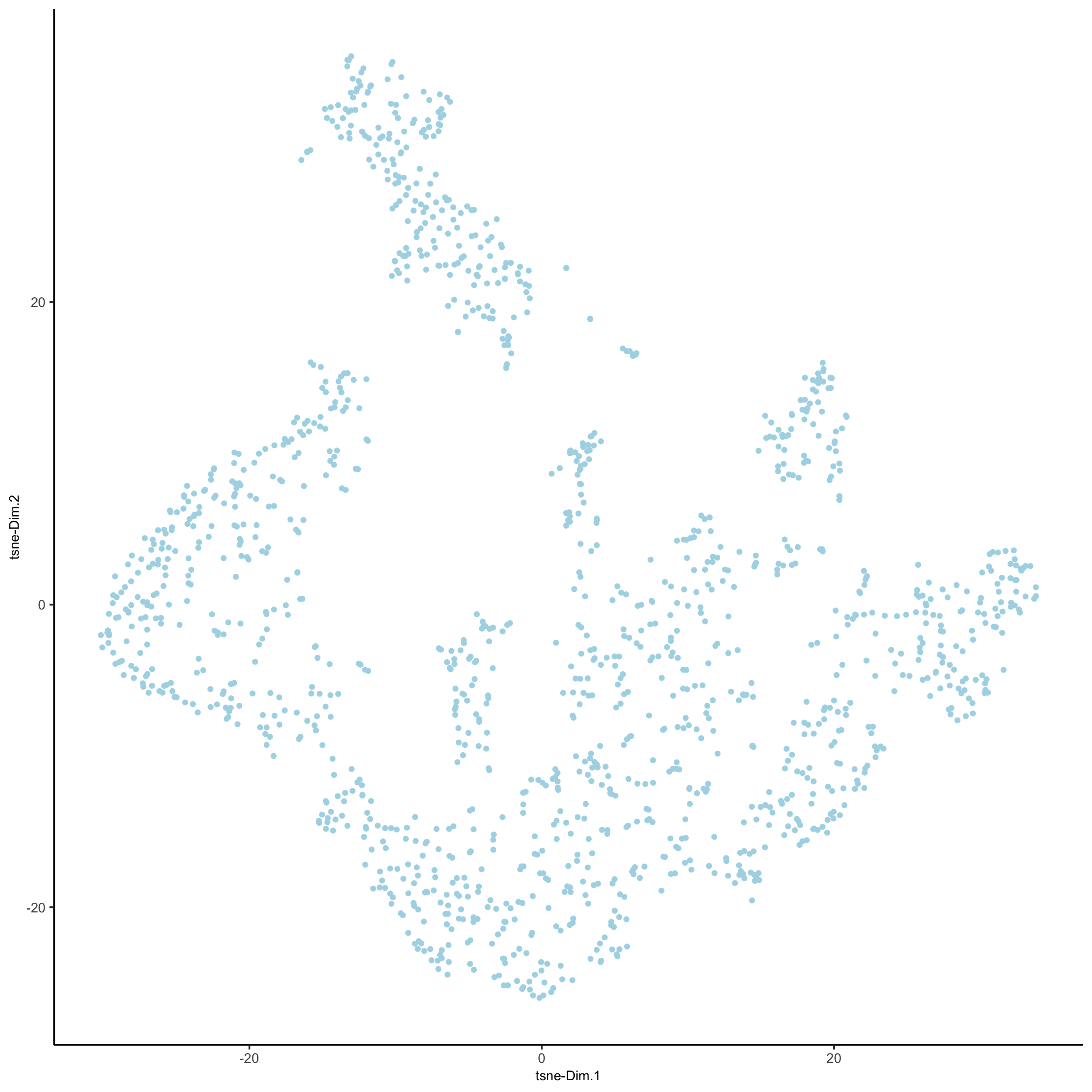
4. Cluster
## sNN network (default)
visium_kidney <- createNearestNetwork(gobject = visium_kidney, dimensions_to_use = 1:10, k = 15)
## Leiden clustering
visium_kidney <- doLeidenCluster(gobject = visium_kidney, resolution = 0.4, n_iterations = 1000)
plotUMAP(gobject = visium_kidney,cell_color = 'leiden_clus', show_NN_network = T, point_size = 2.5,save_param = list(save_name = '4_a_UMAP_leiden'))
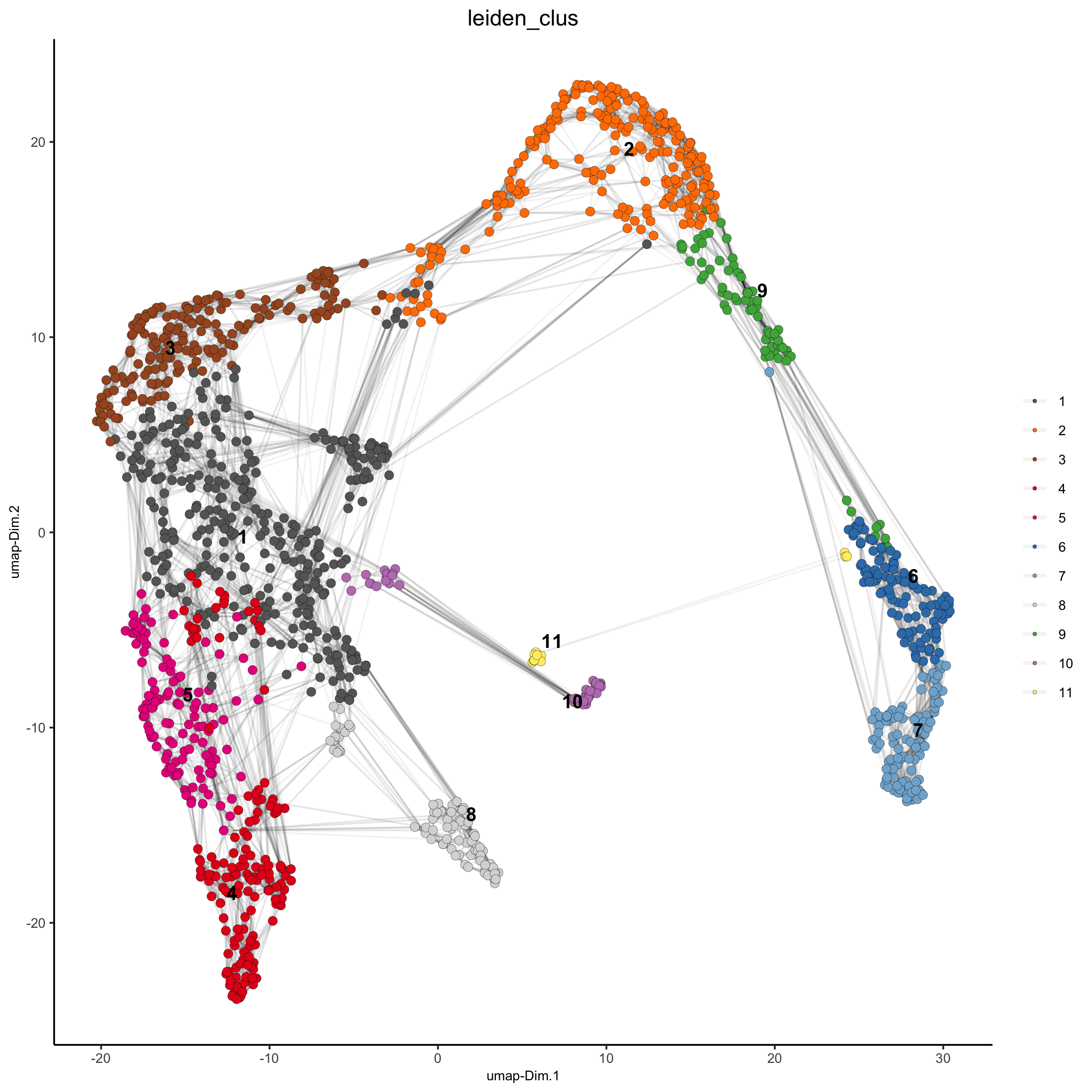
5. Co-visualize
# expression and spatial
spatDimPlot(gobject = visium_kidney, cell_color = 'leiden_clus',dim_point_size = 2, spat_point_size = 2.5,save_param = list(save_name = '5_a_covis_leiden'))
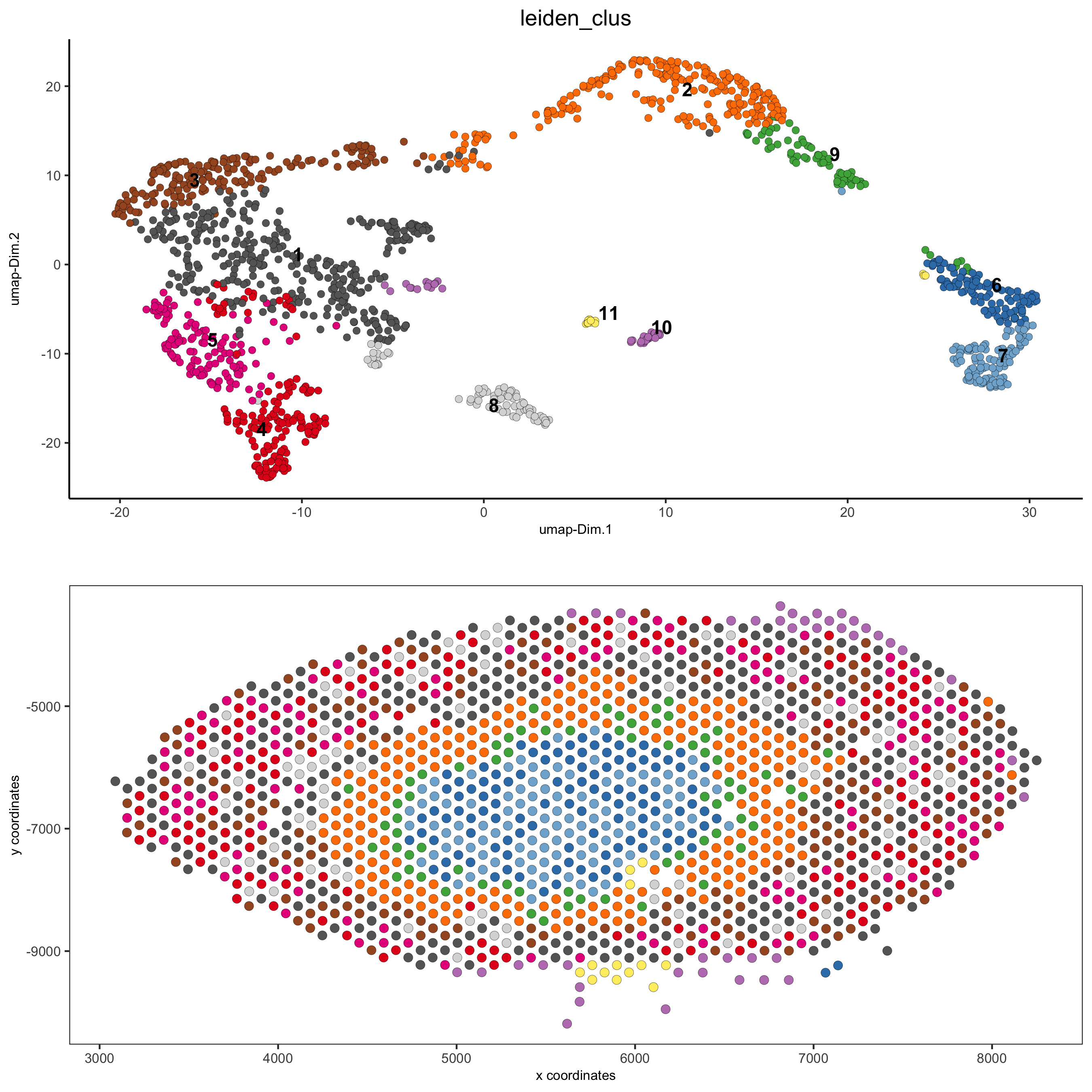
spatDimPlot(gobject = visium_kidney, cell_color = 'nr_genes', color_as_factor = F,dim_point_size = 2, spat_point_size = 2.5,save_param = list(save_name = '5_b_nr_genes'))
6. Cell type marker gene detection
6.1. Gini
gini_markers_subclusters = findMarkers_one_vs_all(gobject = visium_kidney,method = 'gini',expression_values = 'normalized',cluster_column = 'leiden_clus',min_genes = 20,min_expr_gini_score = 0.5,min_det_gini_score = 0.5)
topgenes_gini = gini_markers_subclusters[, head(.SD, 2), by = 'cluster']$genes
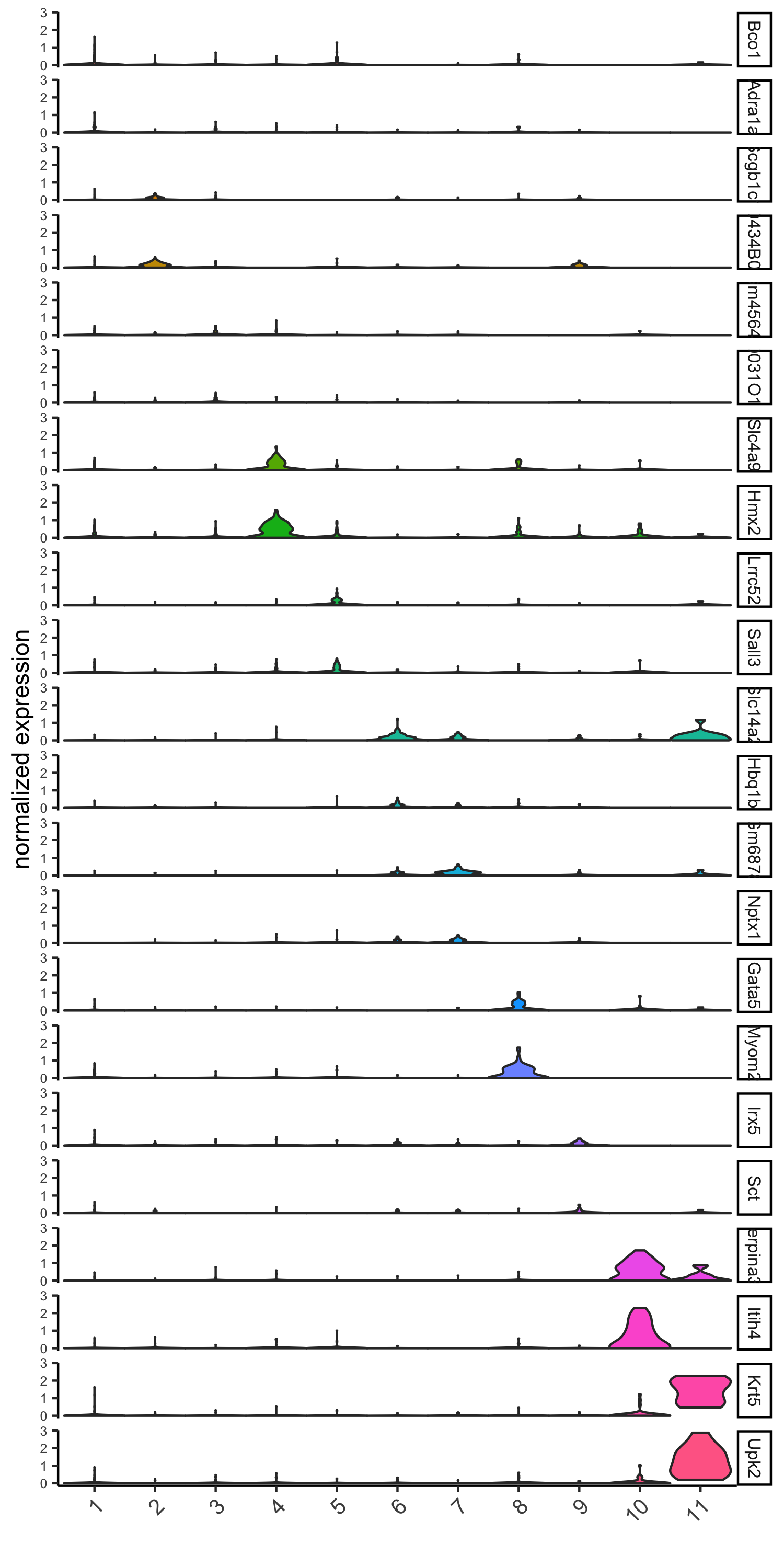
# violinplot
violinPlot(visium_kidney, genes = unique(topgenes_gini), cluster_column = 'leiden_clus',strip_text = 8, strip_position = 'right',save_param = c(save_name = '6_a_violinplot_gini', base_width = 5, base_height = 10))
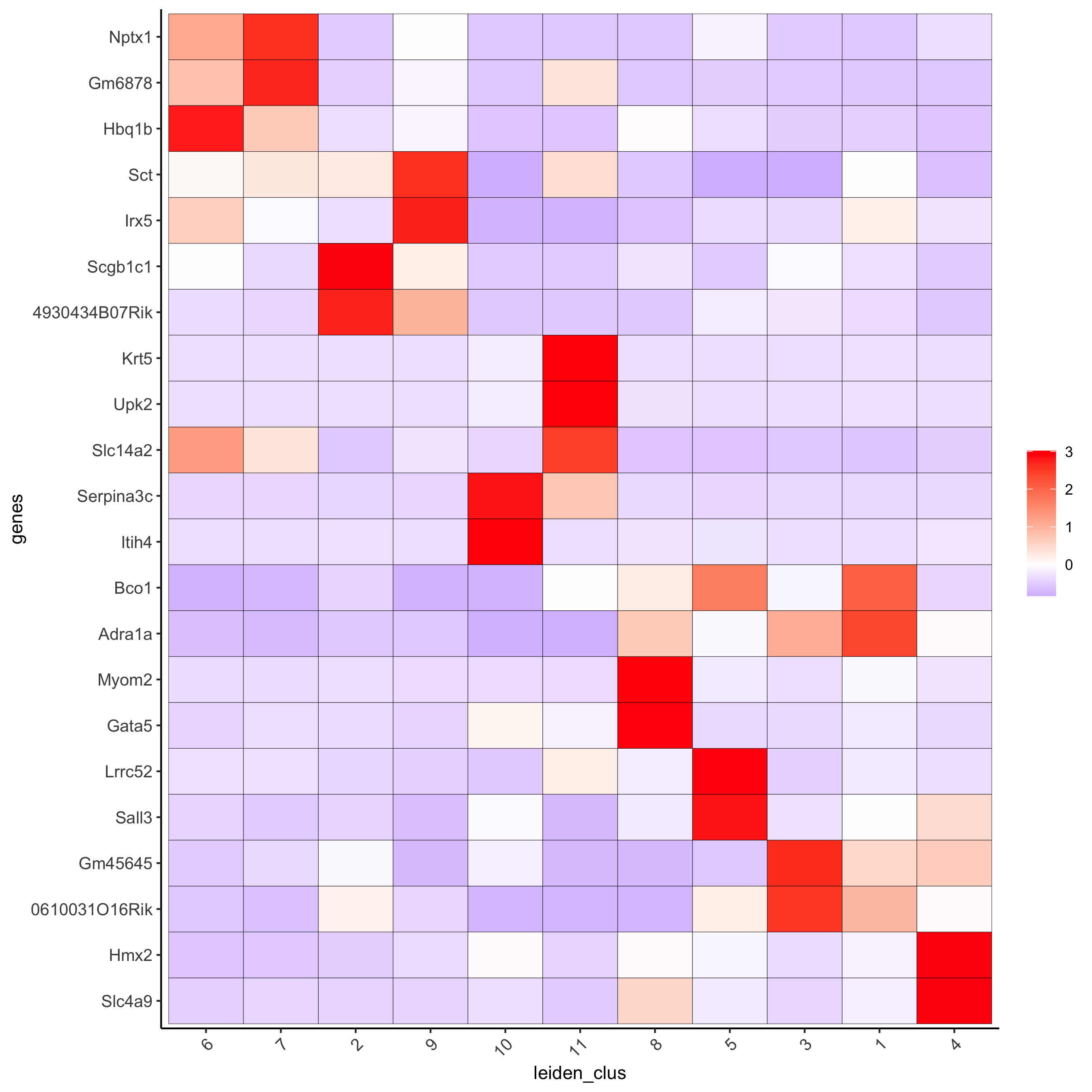
# cluster heatmap
plotMetaDataHeatmap(visium_kidney, selected_genes = topgenes_gini,metadata_cols = c('leiden_clus'),
x_text_size = 10, y_text_size = 10,save_param = c(save_name = '6_b_metaheatmap_gini'))
# umap plots
dimGenePlot2D(visium_kidney, expression_values = 'scaled',genes = gini_markers_subclusters[, head(.SD, 1), by = 'cluster']$genes,cow_n_col = 3, point_size = 1,save_param = c(save_name = '6_c_gini_umap', base_width = 8, base_height = 5))
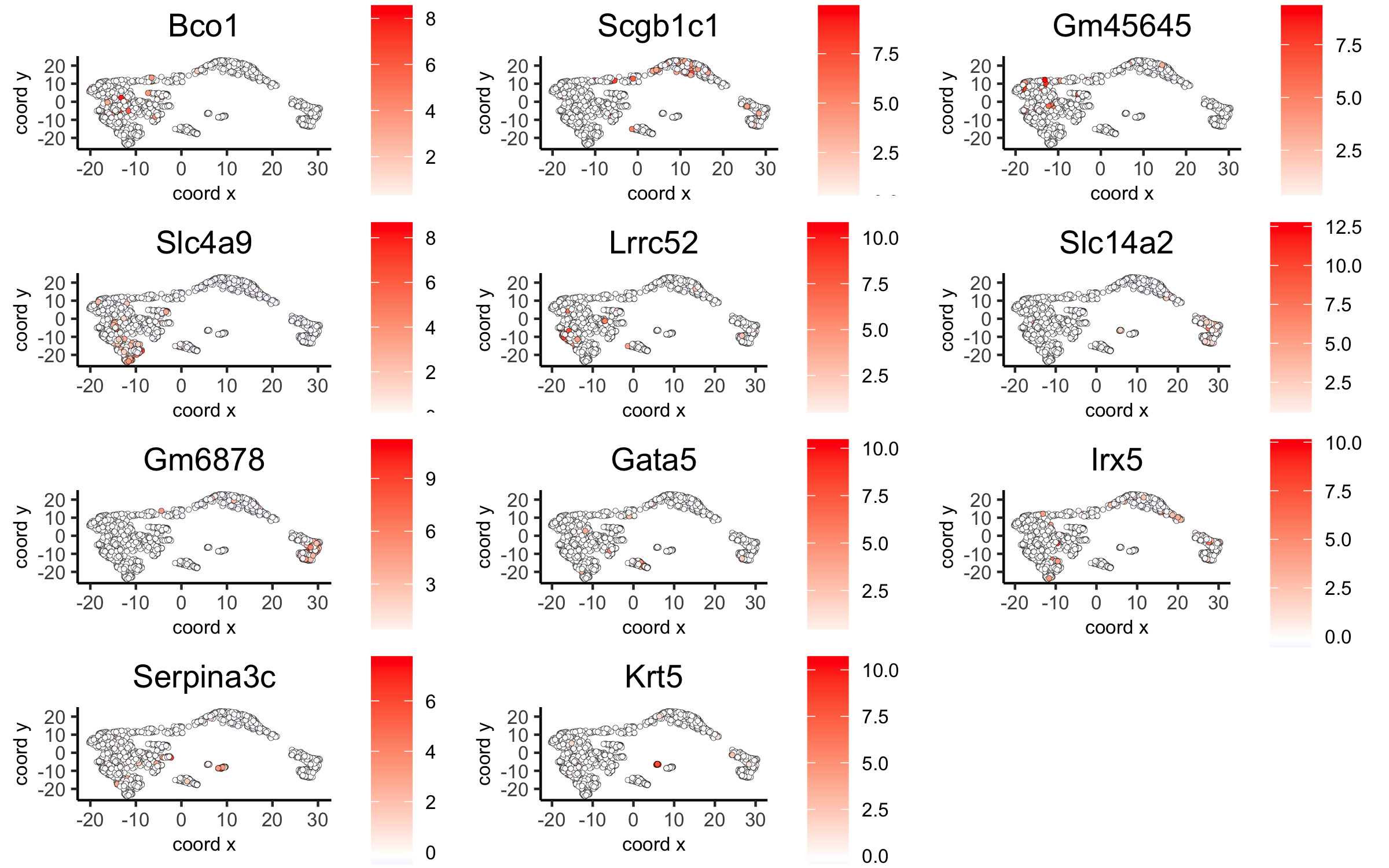
6.2. Scran
scran_markers_subclusters = findMarkers_one_vs_all(gobject = visium_kidney,method = 'scran',expression_values = 'normalized',cluster_column = 'leiden_clus')
topgenes_scran = scran_markers_subclusters[, head(.SD, 2), by = 'cluster']$genes
# violinplot
violinPlot(visium_kidney, genes = unique(topgenes_scran), cluster_column = 'leiden_clus',strip_text = 10, strip_position = 'right',save_param = c(save_name = '6_d_violinplot_scran', base_width = 5))
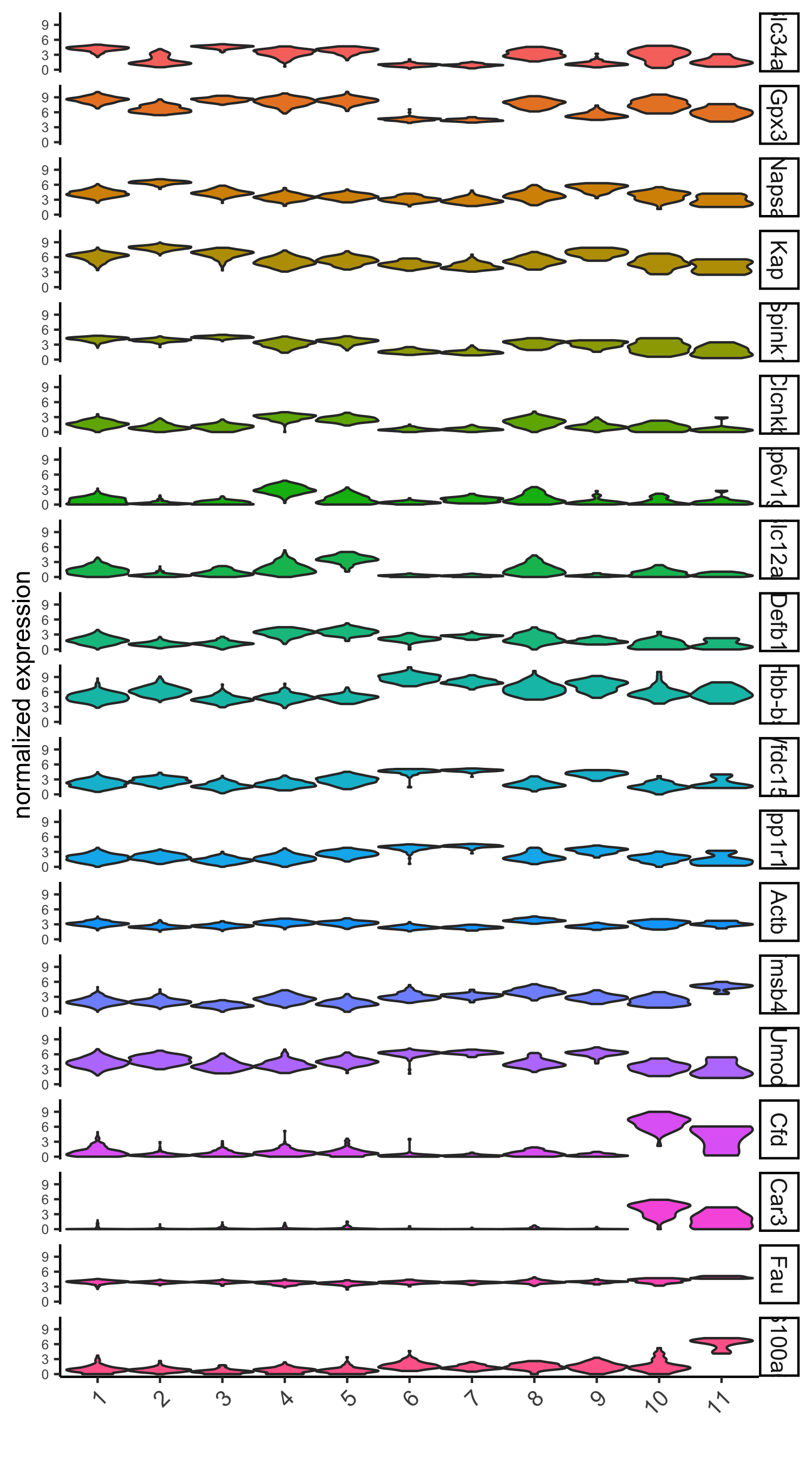
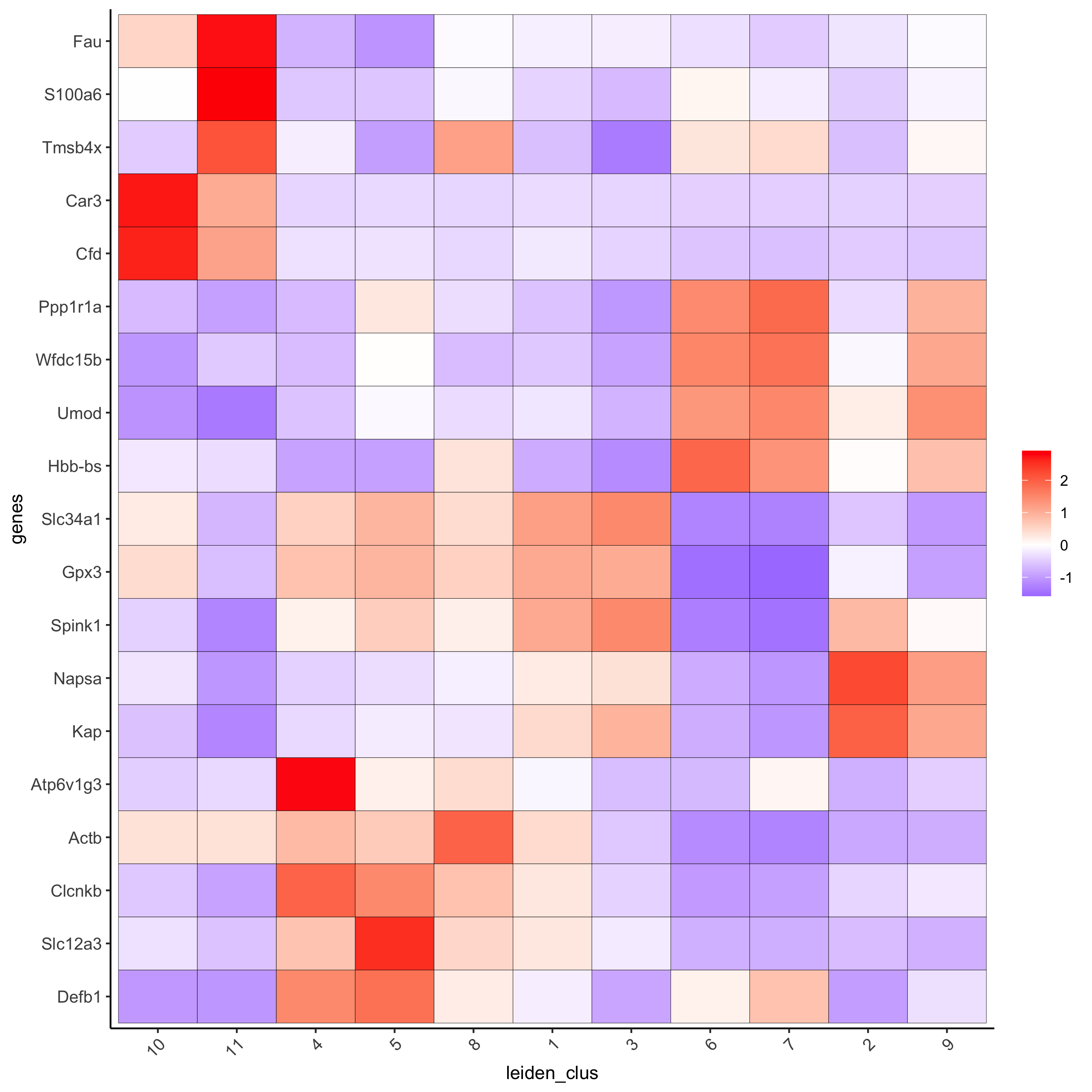
# cluster heatmap
plotMetaDataHeatmap(visium_kidney, selected_genes = topgenes_scran,metadata_cols = c('leiden_clus'),save_param = c(save_name = '6_e_metaheatmap_scran'))
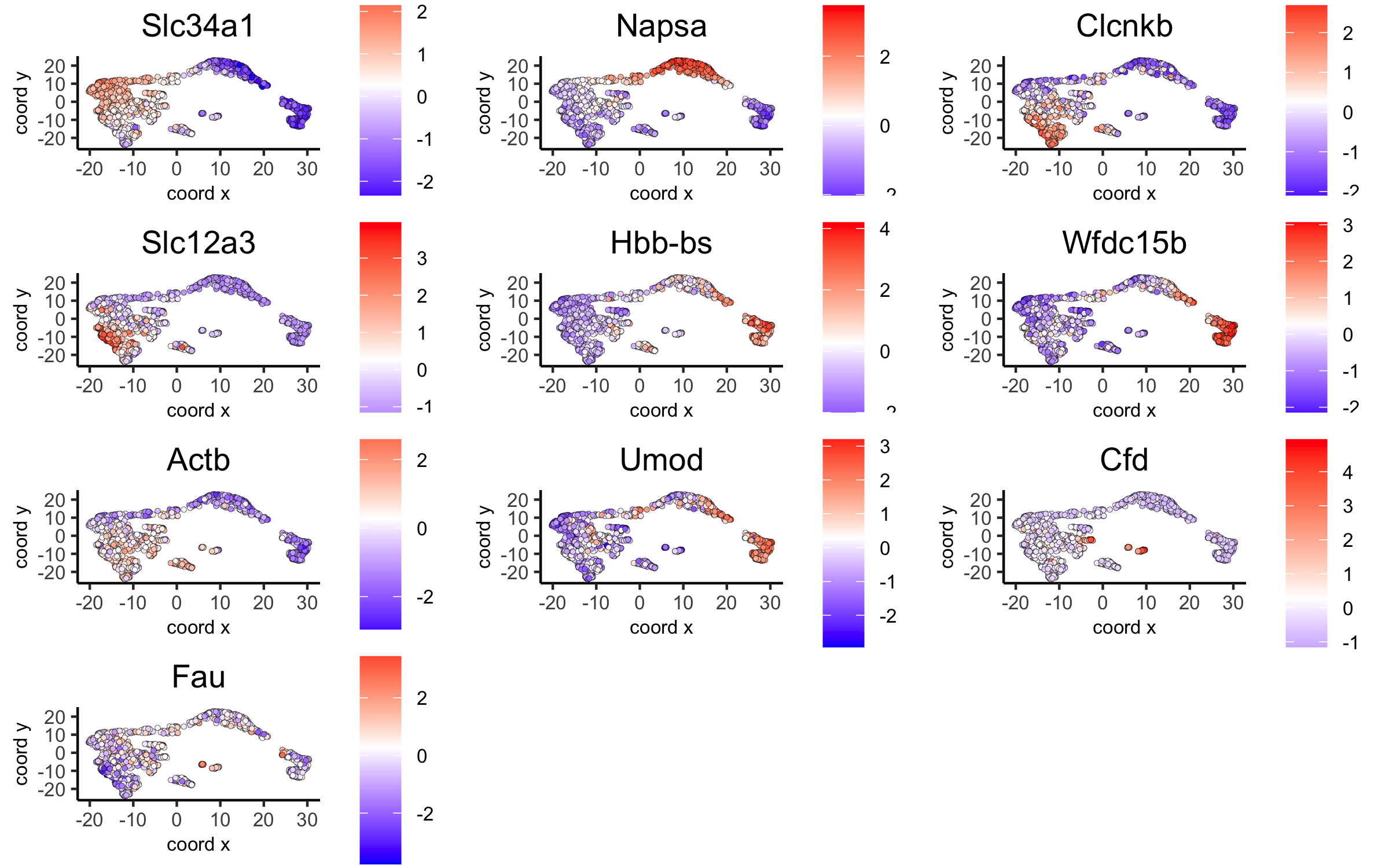
# umap plots
dimGenePlot(visium_kidney, expression_values = 'scaled',genes = scran_markers_subclusters[, head(.SD, 1), by = 'cluster']$genes,cow_n_col = 3, point_size = 1,save_param = c(save_name = '6_f_scran_umap', base_width = 8, base_height = 5))
7. Cell-type annotation
Visium spatial transcriptomics does not provide single-cell resolution, making cell type annotation a harder problem. Giotto provides 3 ways to calculate enrichment of specific cell-type signature gene list:
- PAGE
- rank
- hypergeometric test
See the mouse Visium brain dataset for an example.
8. Spatial grid
visium_kidney <- createSpatialGrid(gobject = visium_kidney,sdimx_stepsize = 400,sdimy_stepsize = 400,minimum_padding = 0)
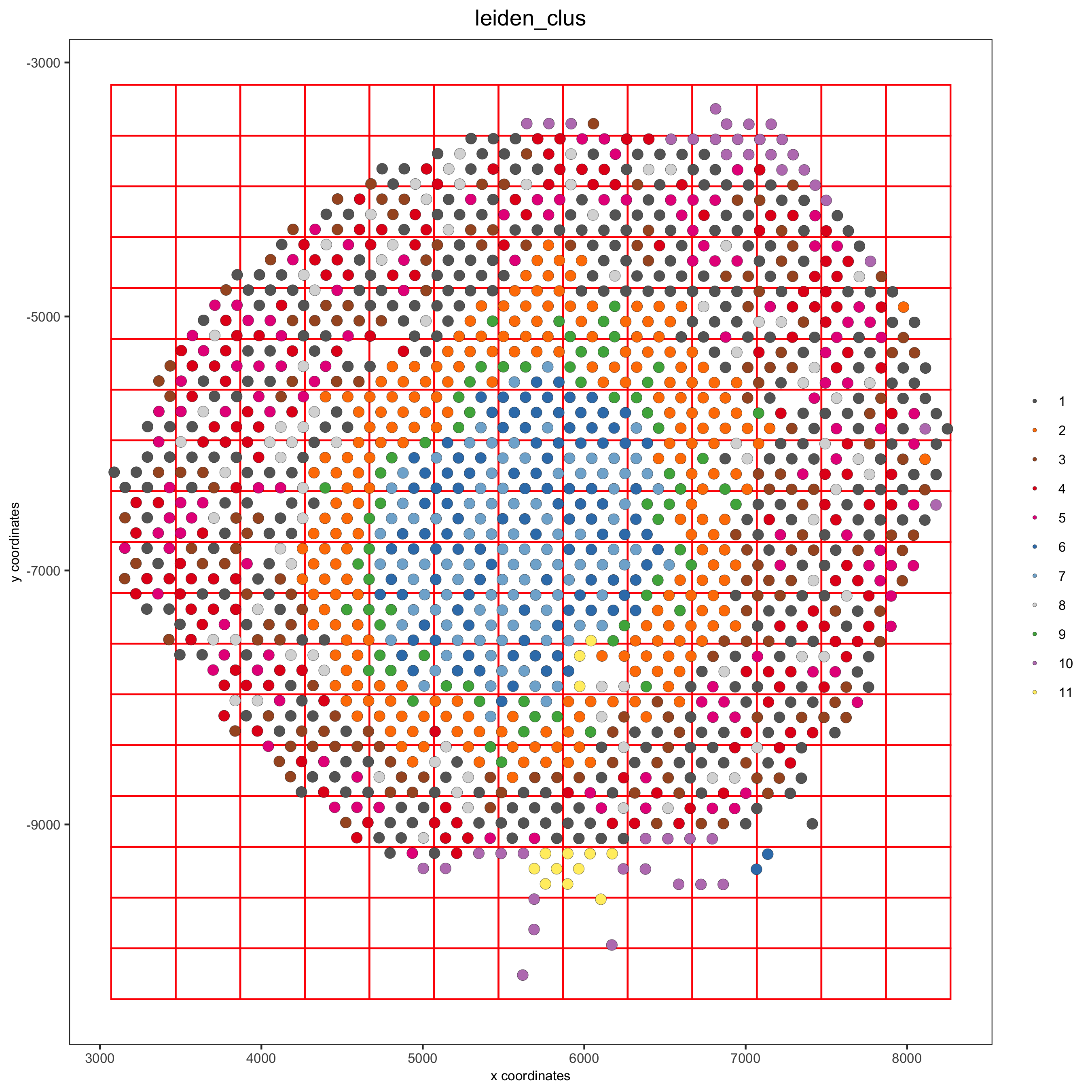
spatPlot(visium_kidney, cell_color = 'leiden_clus', show_grid = T,grid_color = 'red', spatial_grid_name = 'spatial_grid',
save_param = c(save_name = '8_grid'))
9. Spatial network
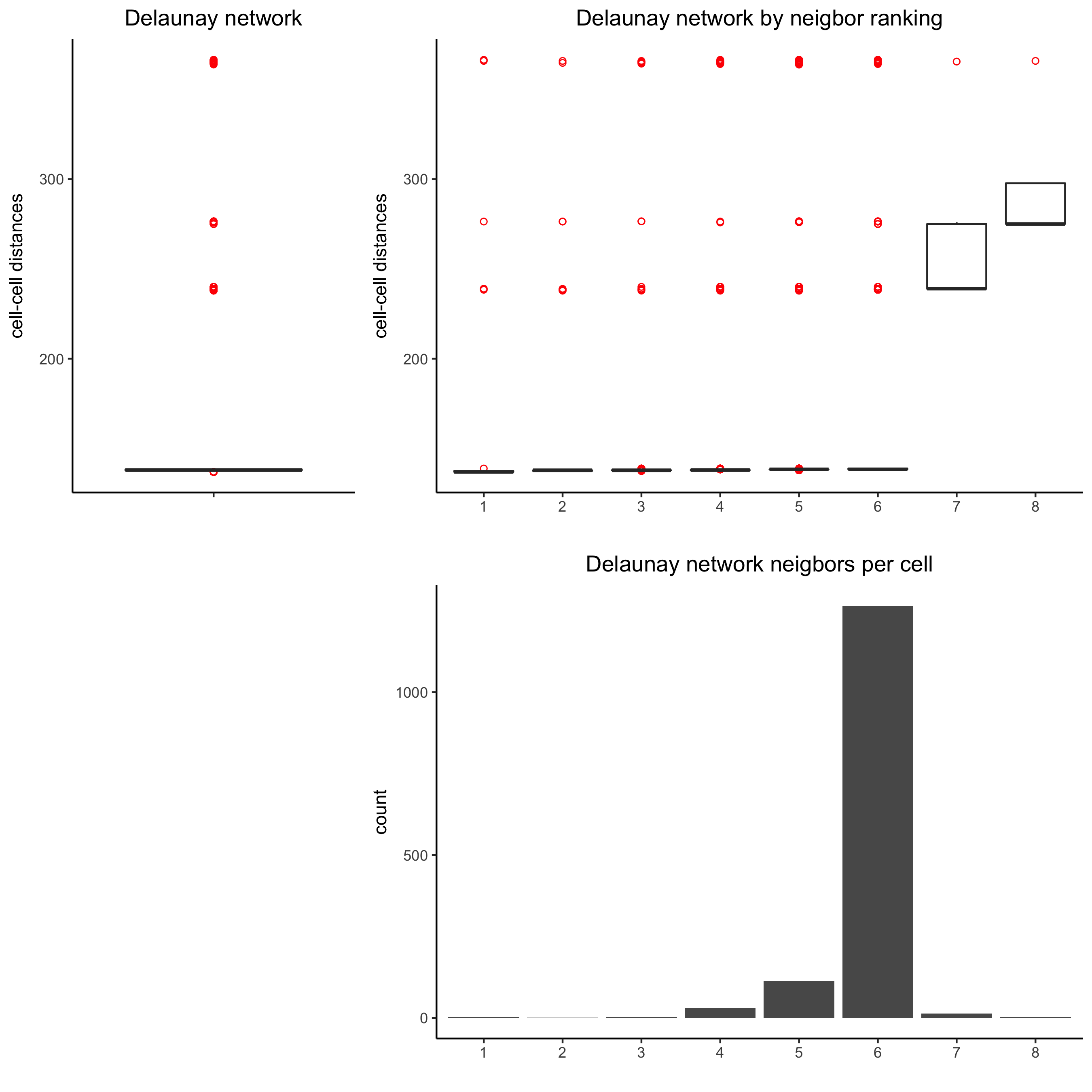
## delaunay network: stats + creation
plotStatDelaunayNetwork(gobject = visium_kidney, maximum_distance = 400,
save_param = c(save_name = '9_a_delaunay_network'))
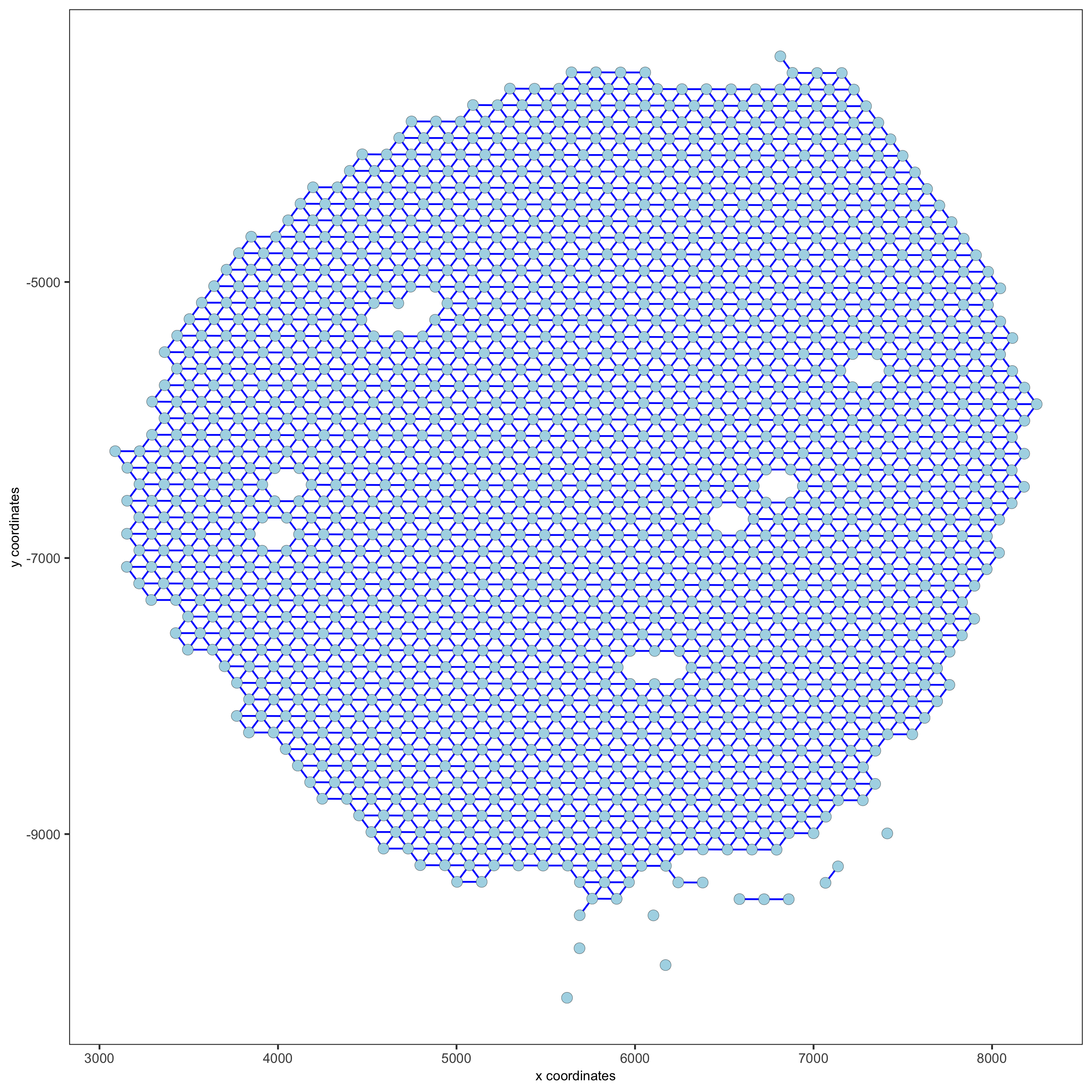
visium_kidney = createSpatialNetwork(gobject = visium_kidney, minimum_k = 0)
showNetworks(visium_kidney)
spatPlot(gobject = visium_kidney, show_network = T,network_color = 'blue', spatial_network_name = 'Delaunay_network',save_param = c(save_name = '9_b_delaunay_network'))
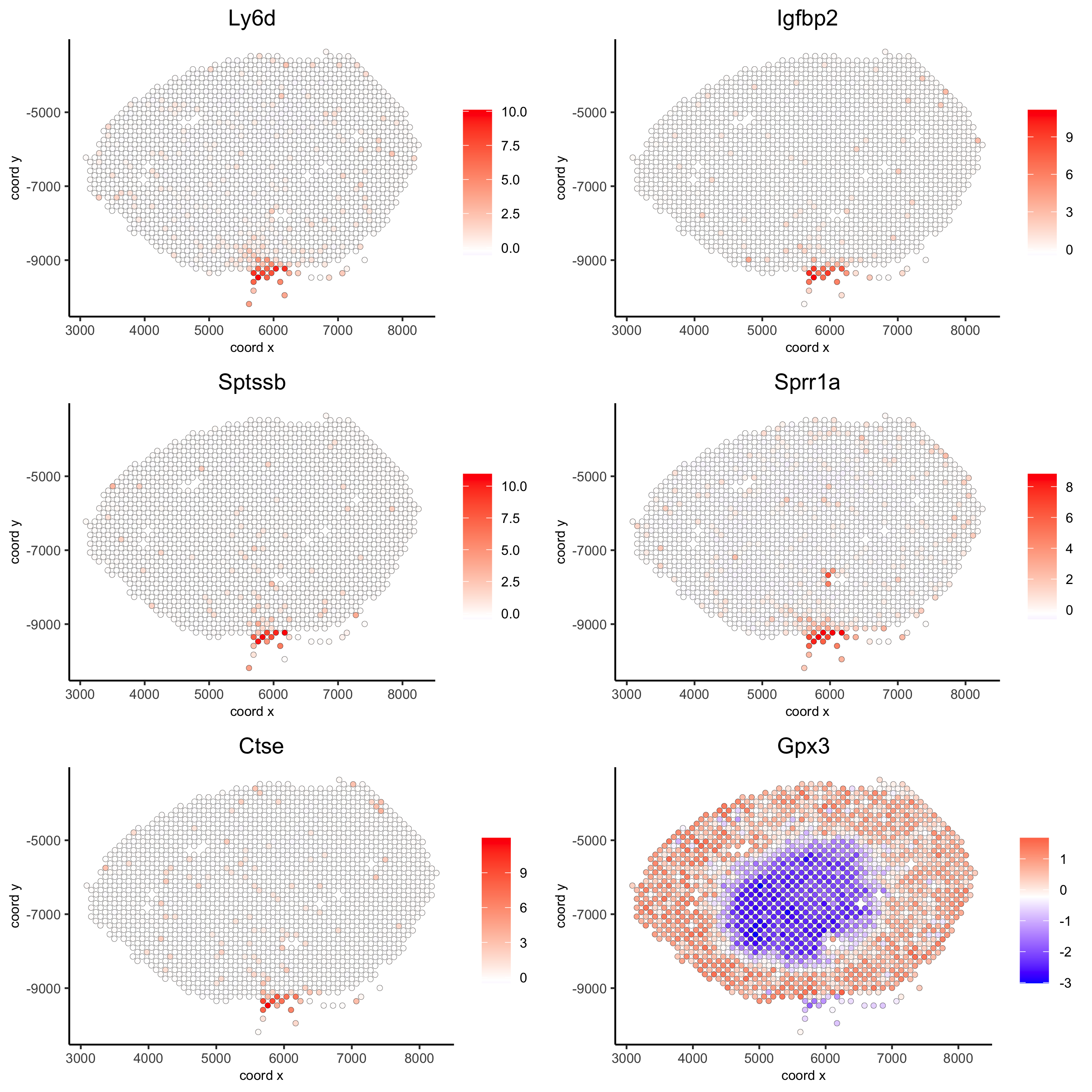
10. Spatial genes
10.1. Spatial genes
## kmeans binarization
kmtest = binSpect(visium_kidney)
spatGenePlot(visium_kidney, expression_values = 'scaled',genes = kmtest$genes[1:6], cow_n_col = 2, point_size = 1.5,save_param = c(save_name = '10_a_spatial_genes_km'))
## rank binarization
ranktest = binSpect(visium_kidney, bin_method = 'rank')
spatGenePlot(visium_kidney, expression_values = 'scaled',genes = ranktest$genes[1:6], cow_n_col = 2, point_size = 1.5,save_param = c(save_name = '10_b_spatial_genes_rank'))
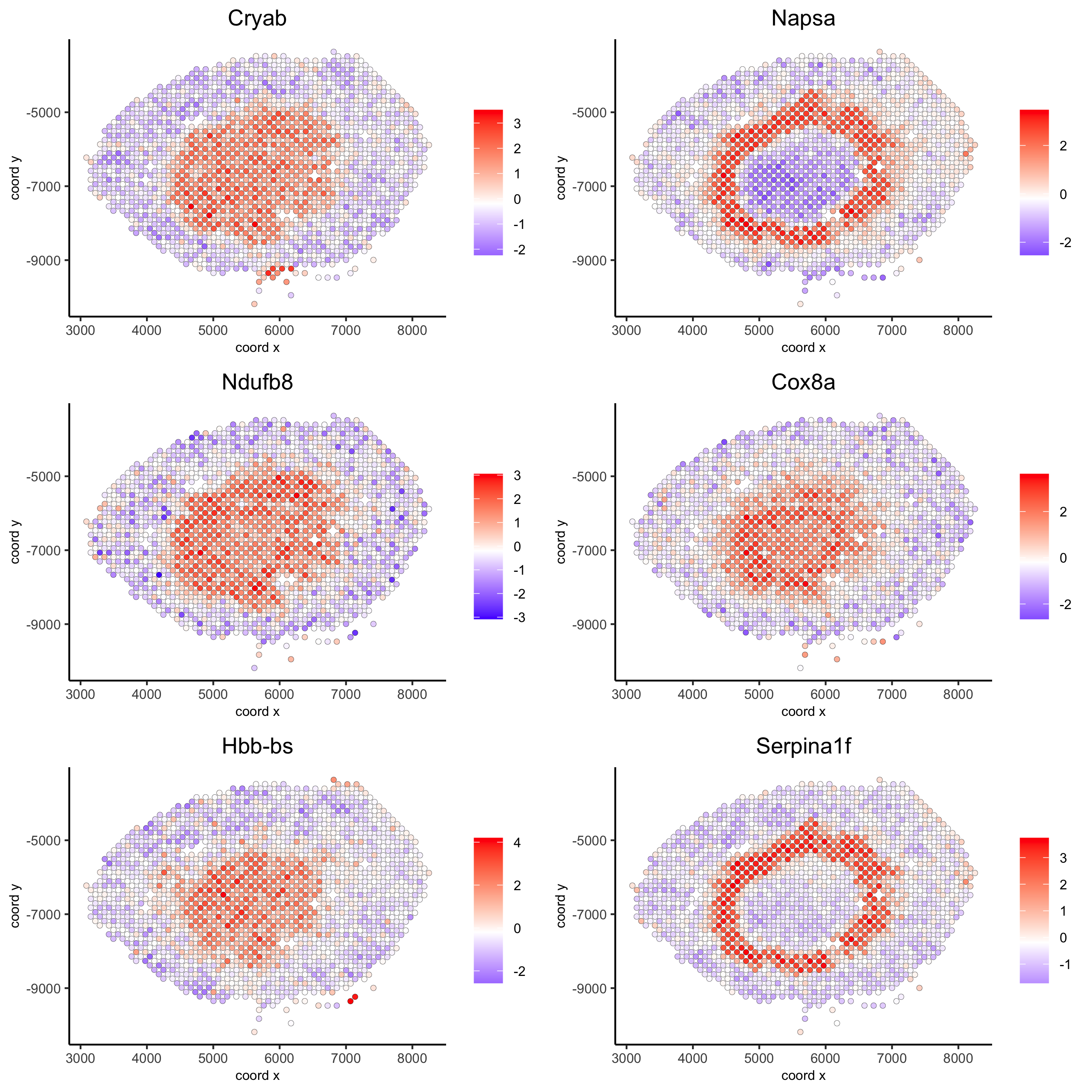
10.2. Spatial co-expression patterns
## spatially correlated genes ##
ext_spatial_genes = kmtest[1:500]$genes
# 1. calculate gene spatial correlation and single-cell correlation
# create spatial correlation object
spat_cor_netw_DT = detectSpatialCorGenes(visium_kidney,
method = 'network',
spatial_network_name = 'Delaunay_network',subset_genes = ext_spatial_genes)
# 2. identify most similar spatially correlated genes for one gene
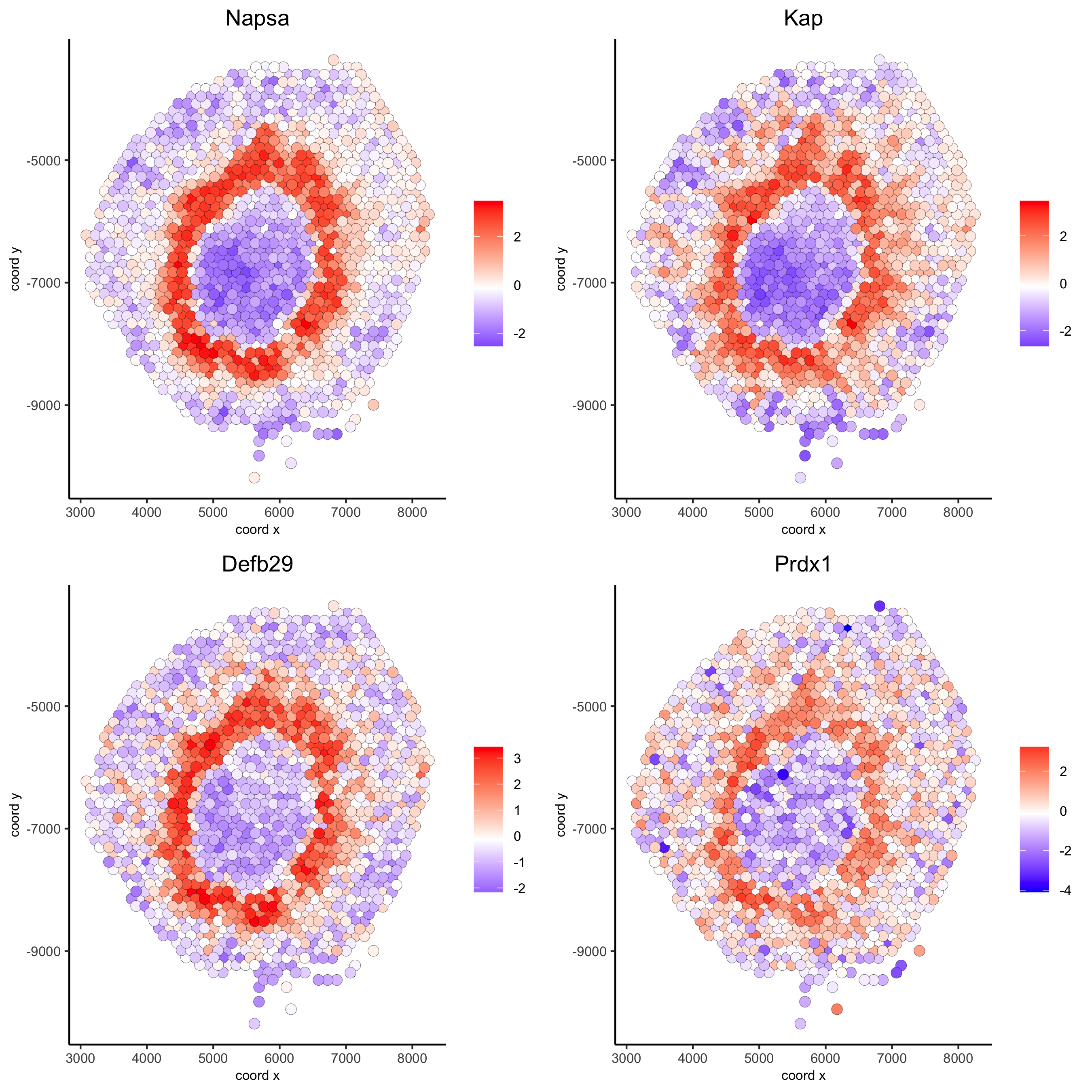
Napsa_top10_genes = showSpatialCorGenes(spat_cor_netw_DT, genes = 'Napsa', show_top_genes = 10)
spatGenePlot(visium_kidney, expression_values = 'scaled',genes = c('Napsa', 'Kap', 'Defb29', 'Prdx1'), point_size = 3,save_param = c(save_name = '10_d_Napsa_correlated_genes'))
# 3. cluster correlated genes & visualize
spat_cor_netw_DT = clusterSpatialCorGenes(spat_cor_netw_DT, name = 'spat_netw_clus', k = 8)
heatmSpatialCorGenes(visium_kidney, spatCorObject = spat_cor_netw_DT, use_clus_name = 'spat_netw_clus',save_param = c(save_name = '10_e_heatmap_correlated_genes', save_format = 'pdf',base_height = 6, base_width = 8, units = 'cm'),
heatmap_legend_param = list(title = NULL))

# 4. rank spatial correlated clusters and show genes for selected clusters
netw_ranks = rankSpatialCorGroups(visium_kidney, spatCorObject = spat_cor_netw_DT, use_clus_name = 'spat_netw_clus',save_param = c(save_name = '10_f_rank_correlated_groups',base_height = 3, base_width = 5))
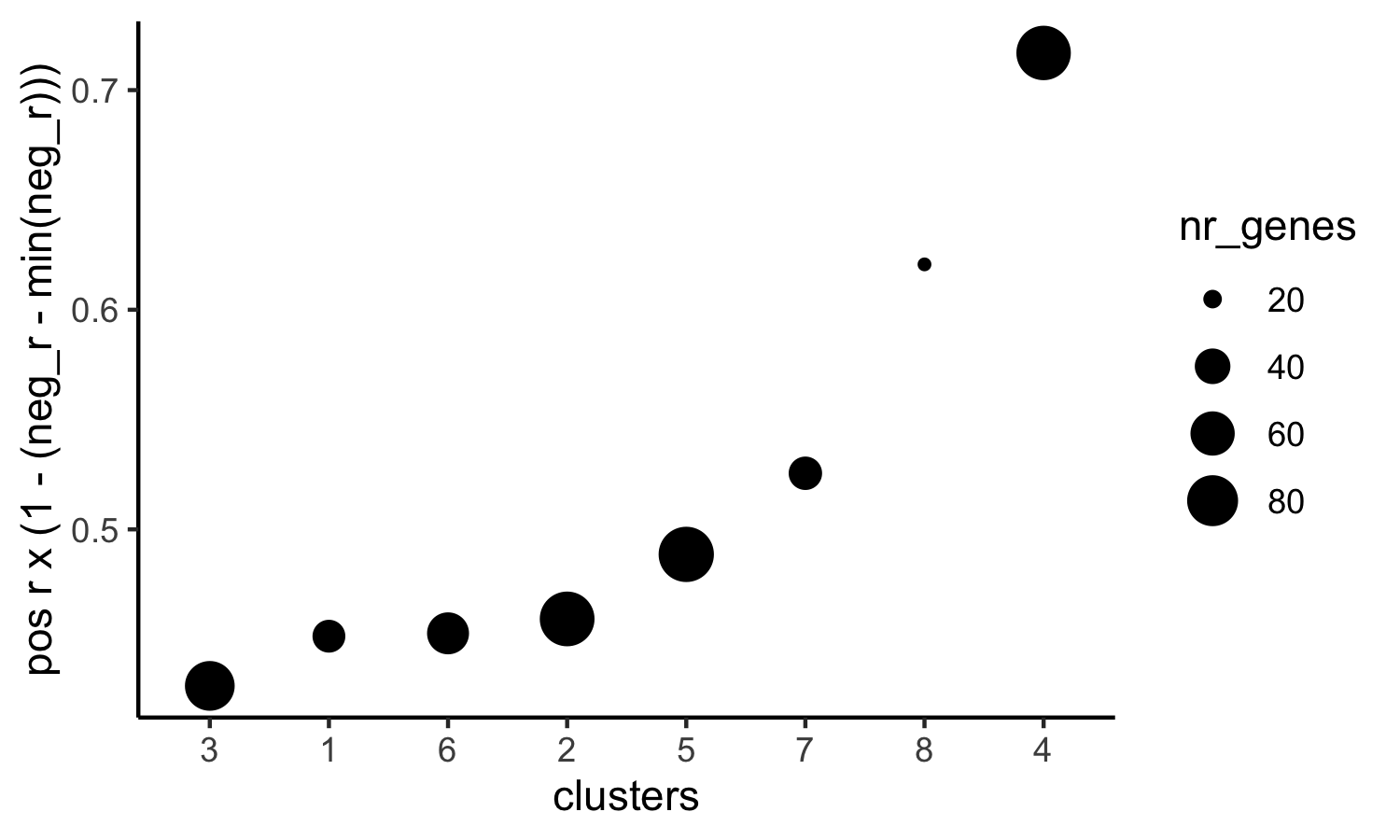
top_netw_spat_cluster = showSpatialCorGenes(spat_cor_netw_DT, use_clus_name = 'spat_netw_clus',selected_clusters = 6, show_top_genes = 1)
# 5. create metagene enrichment score for clusters
cluster_genes_DT = showSpatialCorGenes(spat_cor_netw_DT, use_clus_name = 'spat_netw_clus', show_top_genes = 1)
cluster_genes = cluster_genes_DT$clus; names(cluster_genes) = cluster_genes_DT$gene_ID
visium_kidney = createMetagenes(visium_kidney, gene_clusters = cluster_genes, name = 'cluster_metagene')
spatCellPlot(visium_kidney,spat_enr_names = 'cluster_metagene',cell_annotation_values = netw_ranks$clusters,point_size = 1.5, cow_n_col = 4,
save_param = c(save_name = '10_g_spat_enrichment_score_plots',base_width = 13, base_height = 6))
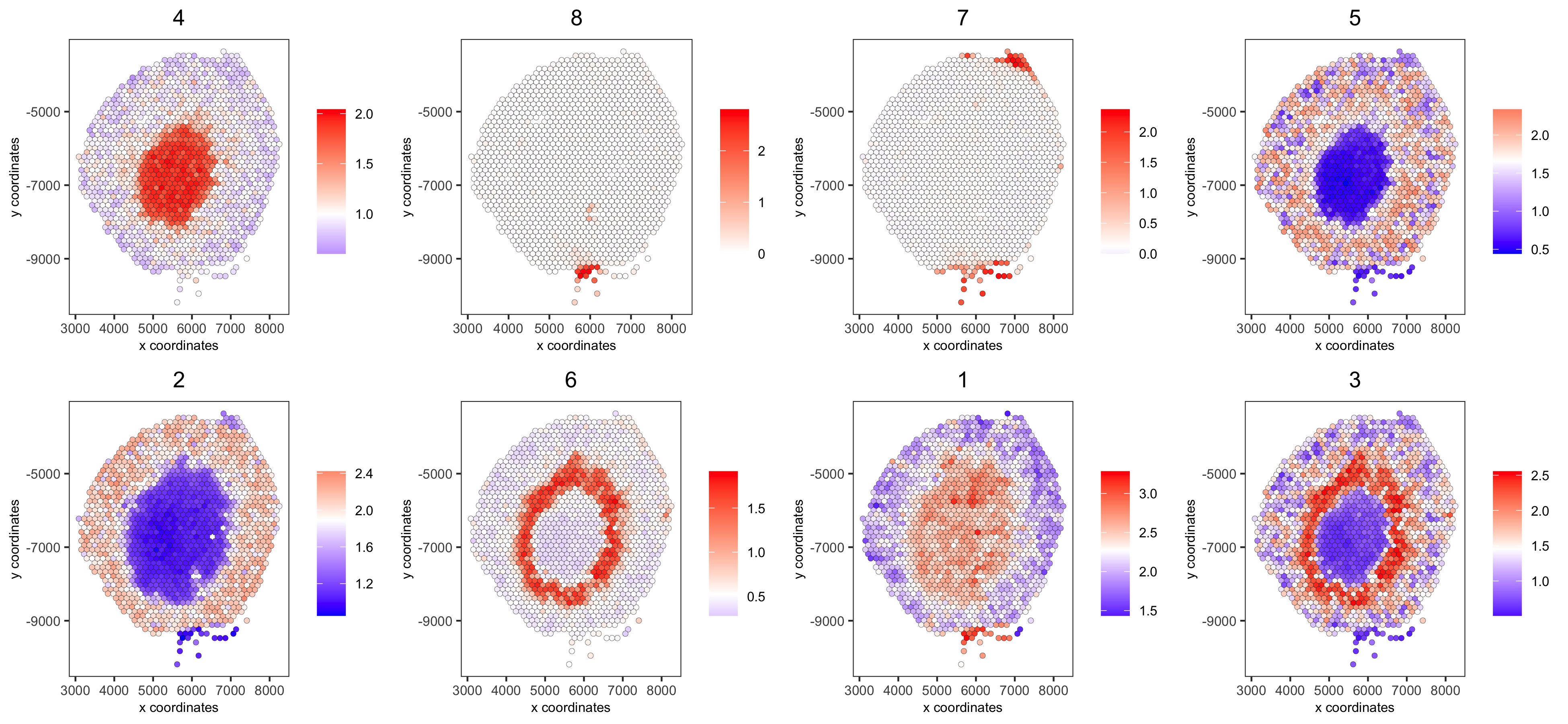
# example for gene per cluster
top_netw_spat_cluster = showSpatialCorGenes(spat_cor_netw_DT, use_clus_name = 'spat_netw_clus',selected_clusters = 1:8, show_top_genes = 1)
first_genes = top_netw_spat_cluster[, head(.SD, 1), by = clus]$gene_ID
cluster_names = top_netw_spat_cluster[, head(.SD, 1), by = clus]$clus
names(first_genes) = cluster_names
first_genes = first_genes[as.character(netw_ranks$clusters)]
spatGenePlot(visium_kidney, genes = first_genes, expression_values = 'scaled', cow_n_col = 4, midpoint = 0, point_size = 2,save_param = c(save_name = '10_h_spat_enrichment_score_plots_genes',base_width = 11, base_height = 6))
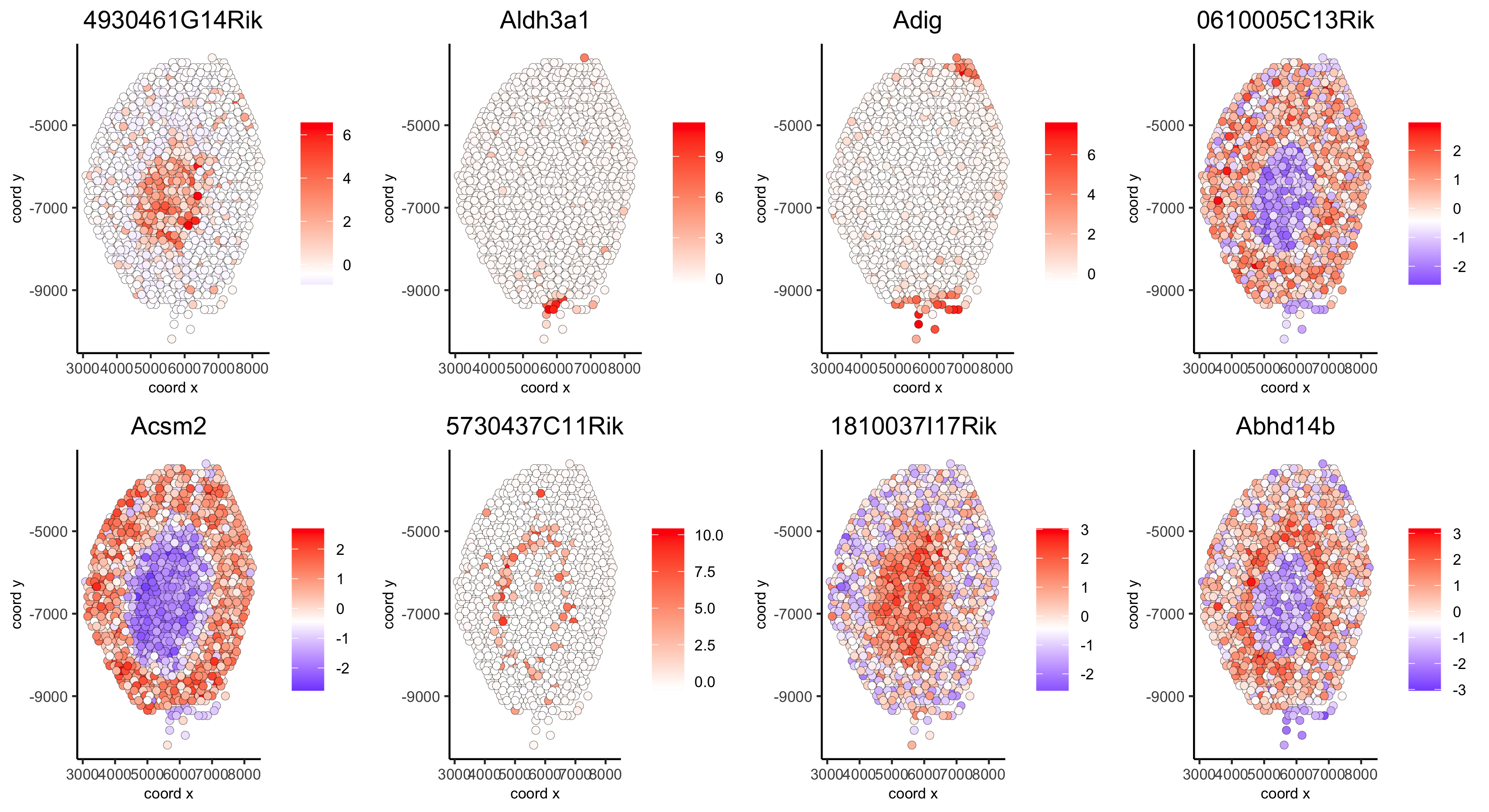
11. HMRF domains
# HMRF requires a fully connected network!
visium_kidney = createSpatialNetwork(gobject = visium_kidney, minimum_k = 2, name = 'Delaunay_full')
# spatial genes
my_spatial_genes <- kmtest[1:100]$genes
# do HMRF with different betas
hmrf_folder = fs::path(my_working_dir,'11_HMRF/')
if(!file.exists(hmrf_folder)) dir.create(hmrf_folder, recursive = T)
HMRF_spatial_genes = doHMRF(gobject = visium_kidney, expression_values = 'scaled',
spatial_network_name = 'Delaunay_full',spatial_genes = my_spatial_genes,k = 5,betas = c(0, 1, 6),
output_folder = fs::path(hmrf_folder, 'Spatial_genes/SG_topgenes_k5_scaled'))
## view results of HMRF
for(i in seq(0, 5, by = 1)) {
viewHMRFresults2D(gobject = visium_kidney,HMRFoutput = HMRF_spatial_genes,k = 5, betas_to_view = i,point_size = 2)
}
## alternative way to view HMRF results
#results = writeHMRFresults(gobject = ST_test,# HMRFoutput = HMRF_spatial_genes,# k = 5, betas_to_view = seq(0, 25, by = 5))
#ST_test = addCellMetadata(ST_test, new_metadata = results, by_column = T, column_cell_ID = 'cell_ID')
## add HMRF of interest to giotto object
visium_kidney = addHMRF(gobject = visium_kidney,HMRFoutput = HMRF_spatial_genes,k = 5, betas_to_add = c(0, 2),hmrf_name = 'HMRF')
## visualize
spatPlot(gobject = visium_kidney, cell_color = 'HMRF_k5_b.0', point_size = 5,save_param = c(save_name = '11_a_HMRF_k5_b.0'))
spatPlot(gobject = visium_kidney, cell_color = 'HMRF_k5_b.2', point_size = 5,save_param = c(save_name = '11_b_HMRF_k5_b.2'))
12. Export and create Giotto Viewer
# check which annotations are available
combineMetadata(visium_kidney)
# select annotations, reductions and expression values to view in Giotto Viewer
viewer_folder = fs::path(my_working_dir, 'mouse_visium_kidney_viewer')
exportGiottoViewer(gobject = visium_kidney,output_directory = viewer_folder,spat_enr_names = 'PAGE',
factor_annotations = c('in_tissue','leiden_clus'),numeric_annotations = c('nr_genes','clus_25'),dim_reductions = c('tsne', 'umap'),dim_reduction_names = c('tsne', 'umap'),expression_values = 'scaled',expression_rounding = 2,overwrite_dir = T)
#================================================================
#Next steps. Please manually run the following in a SHELL terminal:
#================================================================
cd /data/mouse_visium_kidney_viewer
giotto_setup_image --require-stitch=n --image=n --image-multi-channel=n --segmentation=n --multi-fov=n --output-json=step1.json
smfish_step1_setup -c step1.json
giotto_setup_viewer --num-panel=2 --input-preprocess-json=step1.json --panel-1=PanelPhysicalSimple --panel-2=PanelTsne --output-json=step2.json --input-annotation-list=annotation_list.txt
smfish_read_config -c step2.json -o test.dec6.js -p test.dec6.html -q test.dec6.css
giotto_copy_js_css --output .
python3 -m http.server
================================================================
#Finally, open your browser, navigate to http://localhost:8000/. Then click on the file test.dec6.html to see the viewer.
Do as directed.
Note this does not display H&E staining image.
There is a version of this that displays staining image. giotto.viewer.setup3.html#mode_with_images, see section Advanced (with image).